有机无机杂化钙钛矿(HOIPs)是近年来备受关注的新一代光伏材料,仅用了10年时间效率就上升到了25.5%。除了进一步提升HOIPs的PCE外,人们还在钙钛矿太阳能电池的稳定性方面做了许多努力,如构建2D/3D混合维钙钛矿、电荷传输层修饰、层间插入、和封装来抑制HOIPs的分解。然而,由于这些工作对材料固有稳定性的影响有限,并且Pb的毒性给钙钛矿电池的发展也带来了困境,采用铅取代法有望彻底消除HOIPs的毒性问题。高德拜温度可以降低太阳能电池的非辐射复合,探索具有无铅和德拜温度高优点的HOIPs对于追求钙钛矿太阳能电池的优异效率和热稳定性具有重要意义。双钙钛矿A2BB'X6以分子阳离子A、金属阳离子B/B '和阴离子桥接配体X为主要配体,由于其电子结构的多样性和材料选择的多样性,已成为一种新型的无铅钙钛矿材料。然而,目前对无铅的研究大多集中在无机材料上,如Cs2AgBiBr6。目前A2BB'X6材料其主要存在以下问题:1)其吸收通常低于650 nm;2) A2BB'X6的导热系数低,导致器件工作温度高;3)结构复杂,难以保持晶格稳定性;4)小极化子或自陷激子的形成限制了迁移率,从而抑制了扩散/漂移长度;5)多种材料选择导致试错法耗时耗力。因此,寻找性能优良、稳定性好的新型A2BB'X6钙钛矿材料已成为一项紧迫的任务。与传统的基于物理化学背景研究者的直觉来发现新材料的试错方法相比,基于密度泛函理论的高通量计算等先进技术的出现大大加快了新材料的探索过程。然而,由于真实世界中化学空间的大尺度以及材料本身的复杂性严重阻碍了其效率。幸运的是,材料基因组计划和人工智能技术的迅速发展为突破这种困境带来了令人振奋的希望。与第一性原理方法不同,机器学习技术一旦建立了合适的材料数据库并选择了有效的模型,就可以平稳快速地预测一个或多个目标的性质,而不依赖于数值求解复杂的量子力学方程组,这需要比传统方法少几个数量级的计算资源。近年来,机器学习技术在合理材料的设计方面取得了显著进展,如光伏材料、催化剂、锂电池等。值得注意的是,许多用机器学习技术预测的材料已经在实验中合成,并表现出优异的性能。这些成功的尝试表明,智能的机器学习技术绕过密集的DFT计算或实验试验,可以低成本、快速、高精度地预测目标材料性能,将大大加快了材料发现。
复旦大学Yiqiang Zhan和Hao Zhang等人在Adv. Sci.上发表了一篇题目为“Discovery of Lead-Free Perovskites for High-Performance Solar Cells via Machine Learning: Ultrabroadband Absorption, Low Radiative Combination, and Enhanced Thermal Conductivities”的文章。作者通过将高通量DFT计算与机器学习技术相结合,开发了一种多步骤材料筛选方案,以加速发现具有热稳定性高的高性能太阳能电池新型无铅HOIDPs。选择钙钛矿的稳定性、禁带宽度和德拜温度作为目标性能,逐步筛选化学空间。为了在全球范围内搜索可能的HOIDPs候选化合物,首先从元素周期表中基于32种有机阳离子的元素组合的完整化学空间中筛选出包含18038种电中性化合物的数据库。然后利用结构稳定的条件筛选出结构不稳定的候选材料。第三,针对多目标多阶段屏幕建立了高精度的机器学习模型,并分析了相关特征对学习目标的重要性。基于机器学习预测的结果,一些类似斜方的无铅HOIDPs候选材料脱颖而出,并选择基于Br和环境友好的候选光采集技术进行进一步的DFT验证。最后,在带隙合适、德拜温度较高的条件下,筛选出了4种具有较高热稳定性的无铅HOIDPs太阳能电池材料,分别是(CH3NH3)2AgAlBr6, (CH3NH3)2AgGaBr6, (CH3NH3)2AglnABr6和(CH2NH6)2AglnBr6)。
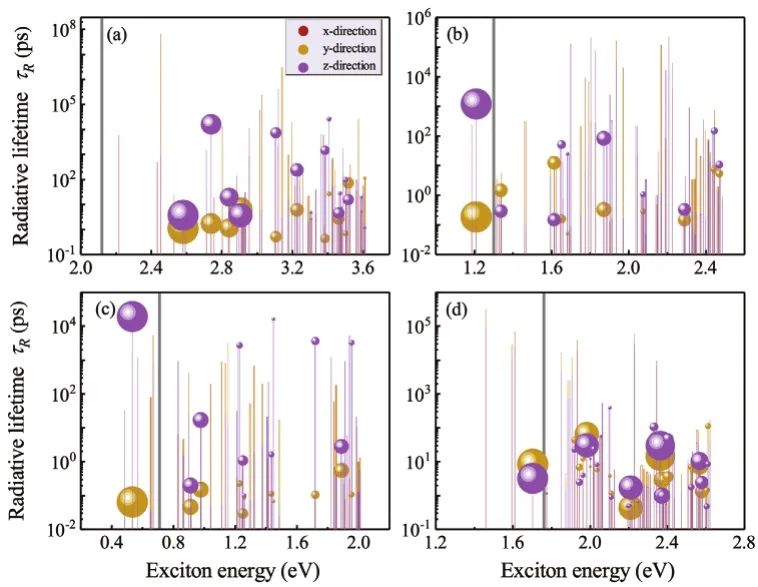
图1左图为光伏应用中结合ML和DFT计算发现新的HOIDPs过程,右图为预测集中钙钛矿的组成和结构。包含32个A位点的单价有机分子阳离子,9个单价,49个二价和35个三价B/B’位点阳离子,以及4个X位点阴离子在元素周期表上的组合产生了一系列尚未探索的HOIDPs候选离子。在电荷中立的条件下,得到了180038个初始候选粒子。然后通过稳定性条件和人工智能法,筛选出了597个适合于太阳能电池的HOIDPs。最后,通过DFT计算进一步验证了这些候选器件的电子特性和其他特性,最终选出了四个质量较好的理想HOIDPs。a) 测试组中每个原子的实际形成能△HDFT和预测的每个原子形成能△HML。通过计算皮尔逊系数(r)、决定系数(R2)、均方误差(MSE)和平均绝对误差(MAE)来估计预测误差。红线表示的理想线表示理想预测结果,黑线表示的拟合线表示实际预测结果。这两条线基本重合。图中所示为预测的△HML和△HDFT的误差百分数。红色曲线显示了趋势。b) 梯度增强回归和SHAP数据库得到的特征重要度排序,元素属性的重要度由高到低。将显示数据集中的所有样本,图中的一个点对应于一个样本。标记为SHAP值的x轴表示特征对地层能量的影响。红色和蓝色分别表示给定特征的高值和低值。c) 化合物摩尔质量下所有电中性候选化合物的生成能的预测。c) GBR模型的R2、MSE和MAE值随所选特征个数的变化。f) 八面体因子的带隙预测关系可视化。不同的颜色代表不同的x位卤素元素。m-p) 室温下5 ps AIMD模拟过程中。筛选的四种HOIDPs ((CH3NH3)2AgAlBr6, (CH3NH3)2AgGaBr6, (CH3NH3)2AglnABr6和(CH2NH6)2AglnBr6)总能量和晶体结构的变化。四种HOIDPs ((CH3NH3)2AgAlBr6, (CH3NH3)2AgGaBr6, (CH3NH3)2AglnABr6和(CH2NH6)2AglnBr6)的光子能量吸收谱。图6 选定HOIDPs多体相互作用诱导的激子寿命。灰线为QP带隙,圆半径表示激子对激子吸收峰的贡献
将高通量DFT计算与人工智能技术相结合,开发了多步材料筛选方案,加速了有机-无机杂化双钙钛矿材料A2BB’X6的发现。从180038种化合物中成功筛选出597种带隙合适、德拜温度高的稳定HOIDP太阳能电池材料。然后,根据无毒和实验可及性条件,又筛选出12种候选材料,其中4种性能优良材料通过了DFT验证。作为新一代材料设计策略,人工智能驱动方案无需深入的物理和化学知识,仅基于现有数据和适当的算法,就可以以“廉价”的方式实现高精度材料发现。同时,人工智能技术可以捕捉隐藏在数据中的结构性质关系,为理解复杂材料性质提供了一种独特的方式,从而帮助研究人员跳出已知知识的框架,找到更合适的描述。此外,多目标筛选可以有效提高筛选效率,且目标数量不受限制。当然,基于人工智能的材料设计方法也存在局限性,如预测精度依赖于数据集的可靠性。此外既要保证数据样本的一致性,又要保证样本的多样性,这也是一个挑战。我们选择了两种模型的预测值都需要满足要求的强制方式,以牺牲最终候选的数量为代价。在未来的研究中可以探索出更灵活的解决方案。文献链接:Discovery of Lead-Free Perovskites for High-Performance Solar Cells via Machine Learning: Ultrabroadband Absorption, Low Radiative Combination, and Enhanced Thermal Conductivities, Adv. Sci., 2021, doi: 10.1002/advs.202103648.







