——1.背景——
基于结构的虚拟筛选是药物发现的重要手段,但是目前许多对接程序都较为耗时,为大规模虚拟筛选造成了一定的挑战。近期,来自MIT以及慕尼黑工业大学的Stärk等人在arxiv上发表了题为《EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction》的文章,提出了基于几何深度学习的方法EquiBind,用于快速确定小分子与蛋白的结合模式,为小分子对接提供了一种全新的解决方案。
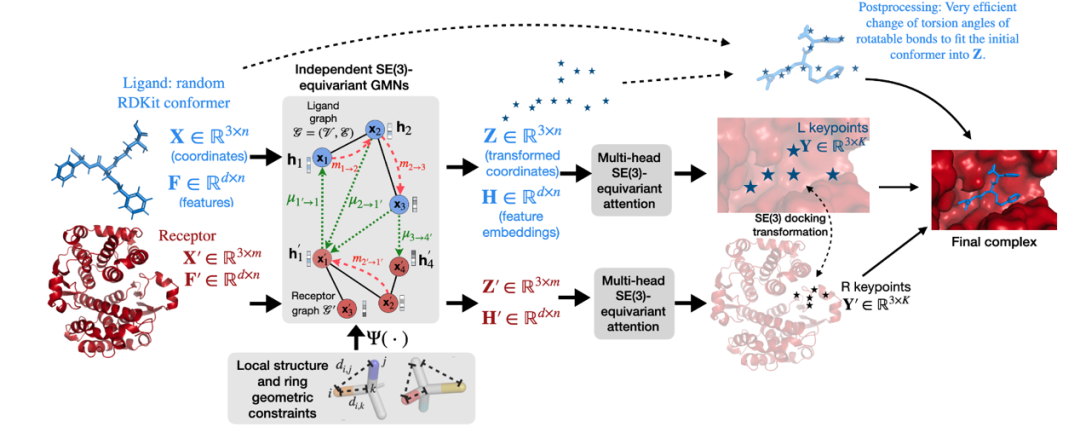
图1:EquiBind的模型架构
图1展示了EquiBind的模型架构。简单来说,该方法利用E(3)协变网络生成配体和受体上的结合关键点,而后在三维空间中对这些点进行匹配来确定结合模式。同时,该网络还能够对配体的三维结构进行变换实现小分子柔性对接。
下面我们简要介绍该模型的架构,并讨论EquiBind在对接上的表现。
——2.方法——
2.1 输入表示
EquiBind程序的输入包括两部分:
· 蛋白质的三维结构(作为刚性输入);
· 由RDKit随机生成的小分子构象(柔性,可优化)。
EquiBind使用K-NN图(k-nearest neighbor graph)来表征这些三维结构,其节点(node)代表原子。对于配体,其节点的特征包括3D坐标及原子类型等。对于蛋白,其节点只包含α-C原子,节点属性为3D坐标以及氨基酸种类。我们在距离接近的节点之间构建边(edge),对于配体和受体,该距离阈值分别为4Å以及30Å。
2.2 E(3)协变网络
在给定输入后,EquiBind需要对输入信息进行分析,并输出蛋白-小分子的结合构象。该任务主要通过一个E(3)协变网络实现,称为IEGMN(Independent E(3)-Equivariant Graph Matching Network),该构架结合了E(3)协变图卷积网络以及图匹配网络,能够在特征提取的同时对3D坐标进行变换。在每一层,模型采取如下信号传递操作:
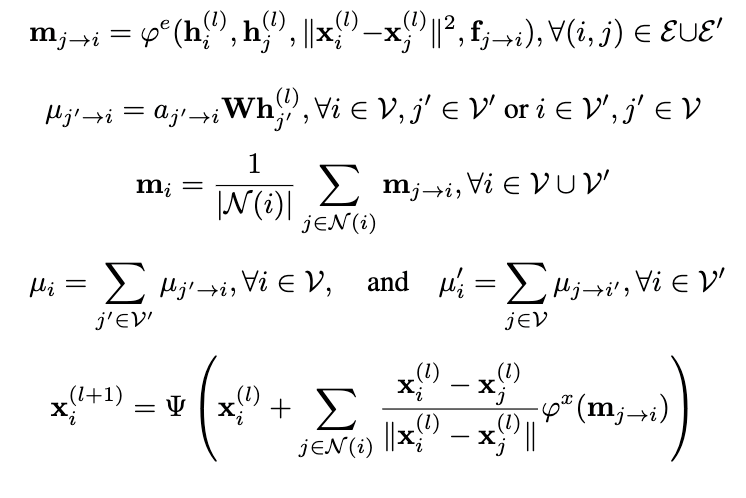
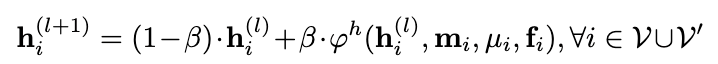
其中hi(l)为节点i在第l层的SE(3)不变表征,xi(l)为节点i在第l层的空间位置。从公式中可以看出,分子图中的节点在每一层中会不断变化。模型在最后一层能够输出其预测的配体结合构象。为了保证配体原子位置的变化符合3D化学规则,EquiBind在每一层会对变换后的坐标进行优化,即公式中的Ψ函数。其具体形式如下:
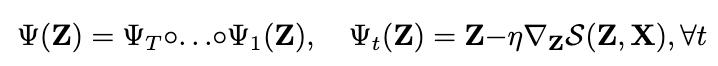
简单的来说,模型利用打分S对配体的三维坐标进行了T步的梯度下降优化。在EquiBind中, S为基于距离的限制函数,其目的是保证分子的局部结构合理。
2.3 结合关键点生成
经过IEGMN处理后,我们得到了经过变换的小分子坐标作为结合构象。同时,EquiBind还会利用IEGMN的输出在蛋白和配体结构中计算结合关键点:
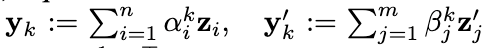
其中zi和zj’分别为小分子和蛋白经过IEGMN变换后的原子坐标,yk以及yk’分别代表经过attention之后的小分子与蛋白内的第k个关键点的位置(关键点的总数是一个固定的超参数,记为K)。在训练过程中,模型通过拟合蛋白-小分子结合界面点的位置来学习yk以及 yk’。上述结合界面点通过计算蛋白-小分子近距离原子(<4Å)的中点得到。由于关键点和界面点的数量一般不一样,EquiBind使用了最优传输损失(Optimal transport loss)对两组点进行匹配从而计算其差异。
利用上述得到的关键点,EquiBind将模型输出的小分子构象叠合至蛋白口袋中,从而完成结合方式的预测。
2.4 小分子结合构象生成
如上文所述,通过IEGMN的三维坐标变换,模型能够预测配体的结合构象。但是IEGMN不能保证输出的3D结构完全正确,在很多情况下其局部结构(如键长和键角)不处于合理范围。为了解决这一问题,EquiBind将RDKit生成的初始构象拟合至IEGMN生成的构象。由于该过程只涉及可旋转单键的变动,因此可以确保局部结构的合理性。其算法主要匹配可旋转单键周围原子位置,不同的可旋转单键可以独立操作,因此该过程可以快速运行。
——3.结果评价——
EquiBind使用PDBBind v2020数据集,其中训练集和验证集包含17,347个复合物结构。EquiBind在测试时使用了PDBBind中发表于2019年后的结构,包含125个蛋白以及363个复合物结构。
作者在评价时主要关注了口袋未知情况下的小分子对接任务。这意味着模型需要找到合适的口袋,然后确定小分子与蛋白的结合方式。评价时用到的baseline包括QVina-W、GNINA、SMINA以及GLIDE。除了标准的运行模式外,作者还比较了EquiBind的其他运行模式,包括:
· EquiBind-U:直接使用IEGMN输出的三维结构计算评价指标(不包含2.4节中提到的构象优化);
· EquiBind-R:将小分子作为刚体进行对接;
· EquiBind-Q:使用QVina 2对EquiBind产生的构象进行优化;
· EquiBind-S:使用SMINA对EquiBind产生的构象进行优化。
作者对以下指标进行了评价:(1)L-RMSD:配体结合pose的RMSD值;(2)Centroid Distance:在真实复合物中配体的中心位置与对接配体中心位置间的距离,可以用于评价模型是否正确确定了口袋的位置;(3)Kabsch RMSD:用于评价小分子内部构象的合理性。
表
1:EquiBind方法评价结果
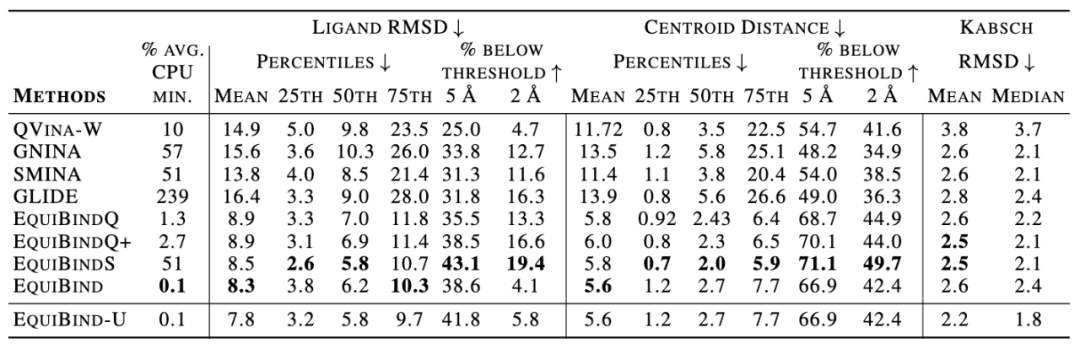
从结果上看,EquiBind在各项指标上均显著优于其他模型。该优势主要体现在Centroid Distance:即EquiBind能够正确找到小分子的结合口袋。同时,EquiBind的运行时间也显著优于其他方法。
——4.总结——
EquiBind是一种全新的基于深度学习的小分子对接方法,能够利用E(3)协变网络快速确定蛋白-小分子的结合方式。该方法为小分子对接提供了一种全新的解决方案,有较好的发展空间。
尽管如此,该方法目前在评价上存在局限性。上述评价过程只涉及结合口袋未知的情况,但是大部分虚拟筛选项目所关注的口袋是确定的。另外,本文所比较的baseline方法主要关注的是口袋已知的对接,在口袋未知的情况下一般表现较差,直接比较可能不公平。后续工作可以评价EquiBind在口袋已知情况下的表现,并与相关baseline进行比较,从而进一步明确其适用范围。
参考文献:
[MAIN] Stärk, Hannes, et al."EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction." arXiv preprintarXiv:2202.05146 (2022).
点击左下角的"阅读原文"即可查看原文章。
作者:李亦博
审稿:王世伟
编辑:由瀚天
GoDesign
ID:Molecular_Design_Lab
( 扫描下方二维码可以订阅哦!)

本文为GoDesign原创编译,如需转载,请在公众号后台留言。







