
摘要
这项研究中,文章提出了一种新颖的多层CNN架构DeepPhos,以准确预测具有蛋白质序列信息的磷酸化位点。与之前深度学习方法不同,DeepPhos由密集连接的CNN(DC-CNN)块组成,其中卷积层通过块内串联层(Intra-BCL)同时相互连接,有效地增强了磷酸化信息的流动。同时,采用具有不同窗口和过滤器尺寸的多个DC-CNN块自动捕获蛋白质磷酸化位点的重要序列表示,并通过块间串联层(Inter-BCL)进一步集成以进行最终预测。为了评估DeepPhos的性能,本文从几个数据库中收集了大量经过验证的磷酸化示例,这些示例用于训练和评估模型。评估结果表明,DeepPhos在一般磷酸化预测方面优于现有方法。此外,该架构可以成功地应用于一系列激酶特异性磷酸化位点预测任务,从激酶基团水平到单个激酶水平,从基础一般DeepPhos模型逐层转移。进一步的评估还表明,DeepPhos在激酶特异性磷酸化位点预测方面具有更好的性能。一、背景
翻译后修饰(PTM)是通过共价和通常酶促修饰调节细胞功能的关键机制,其在调节各种生物过程中起着至关重要的作用。有证据表明,超过30%的真核蛋白可以被磷酸化,其中一半与不同类型的疾病密切相关,特别是癌症。磷酸化是研究最多的翻译后修饰,它对多种生物过程至关重要。近年来,人们开始努力开发磷酸化位点预测的计算预测工具,但大多数都是基于特征选择和鉴别分类。因此,开发一种新的、高度准确的预测器是有用的,可以揭示复杂的蛋白质磷酸化位点。
二、数据收集和预处理
为了确保高质量的数据,本文从多个数据库中收集了经过实验验证的人类蛋白质磷酸化位点。一般的位点预测:从不同的数据库中删除了所有重复系列,然后使用相似性阈值为40%的CD-HIT工具,以减少磷酸化蛋白的序列冗余并避免模型过度拟合。最后,保留了12810个蛋白质序列,提取了所有实验验证的S / T位点和Y位点的磷酸化位点作为正样本,S / T位点和Y位点的数量分别为140 120和27 691。负样本为随机选择的其他S / T和Y位点的子集,与正样本数量相同。对于激酶特异性预测:使用类似的预处理程序,然后本文将所有具有激酶注释的8130个磷酸化位点聚类为组,家族,亚科和单个激酶水平。本研究采用了深度学习方法中常用的性能评估策略进行序列分析,将数据集随机分为严格不重叠的训练,验证和独立测试集。
三、Deepphos框架
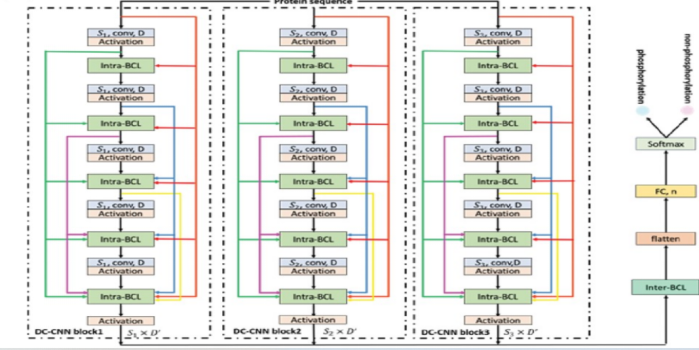
图1.deepphos框架
DeepPhos是一种新的CNN结构,它可以通过一系列非线性变换将局部蛋白序列映射到高维连续表示,最终生成磷酸化位点的分类结果。DeepPhos不是直接使用普通的多层CNN,而是利用不同的DC-CCNN块(图1),有效地确保磷酸化预测的关键蛋白序列信息。对于一个局部蛋白质序列x,完全为K个DC-CNN块的DeepPhos的输入是一组序列特征Ek∈RLk*I,I和Lk分别为氨基酸符号字典的大小和相应的磷酸化位点的局部窗口大小。在本研究中,通过one-hot编码方案对蛋白质序列进行编码,因此I被设置为21。本文仔细探索了各种配置的DC-CNN块与不同的窗口大小在磷酸化网站预测的任务,最后开发一个高效的网络架构,K = 3 ,窗口大小为15,33和51。每个DC-CNN块中的卷积层沿着蛋白质序列长度进行连续卷积操作,生成相应的值,然后应用于激活函数ak(本文使用ReLU),以激活神经元,实现非线性变换。对于DC-CNN块k,由第一个卷积层生成的特征图定义为:
其中wk表示大小为的权重矩阵,bk1表示偏置项。为了减少训练中过拟合的风险,每个卷积层都采用dropout,在卷积层后随机丢弃部分神经元。为了增强DeepPhos的DCCNN块中的磷酸化信息流,引入了intra-bcl块,它将所有之前的卷积层与随后的卷积层连接起来,将不同层次的前一层的抽象转移到当前层,增加网络的深度。因此,将DC-CNN块k中第i个卷积层的输出特征图沿特征维数连接,计算方法如下:
其中bki-1是指在i-1个卷积层中生成的特征图,,D'是卷积层1到i的滤波器总数,C是每个DC-CNN块中的卷积层数,在本研究中设置为5。在使用不同的DC-CNN块生成蛋白质磷酸化位点的序列表示后,它们通过DeepPhos中的Inter-BCL进一步整合,沿着第一个维度执行连接如下: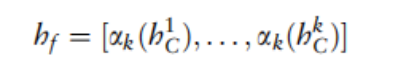
bkc表示第k个DC-CNN块中的卷积层C生成的特征图。这样,将多个特征映射连接起来,然后通过一个flatten层转换为一维张量。然后应用全连接的神经网络生成最终的softmax函数的输入。最后,计算出磷酸化的预测评分如下: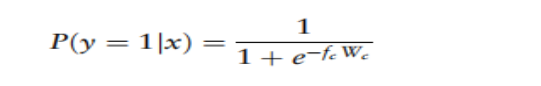
DeepPhos可用于磷酸化位点预测,包括组、家族、亚科或个体激酶水平的一般和激酶特异性预测。为此,对于一般的磷酸化位点预测,所有可用的S/T和Y磷酸化位点数据都被用于训练深度学习模型。另一方面,深度学习模型对激酶特异性磷酸化位点预测的训练更具挑战性,因为目前大多数已验证的磷酸化位点都缺乏相应的激酶注释。为了解决这个问题,本文首先通过不需要激酶注释的磷酸化数据来训练和验证了一个深度学习模型Mp。然后,本文利用激酶特异性的训练和验证数据,进一步微调Mp,获得最终的深度学习模型Mt。在本研究中,本文采用了迁移学习微调策略,在Mp中转移网络,包括所有DC-CNN块的卷积层、intra-BCL块的卷积层,以及与卷积层相关的学习权重矩阵和偏差项。四、性能比较
本文首先比较了DeepPhos与不同的深度学习网络架构,包括CNN、RNN、全连接神经网络(FCNN)和LSTM。这些方法对S/T和Y的AUC值列于表1。总的来说,DeepPhos比其他深度学习架构获得了更高的AUC值,这表明DeepPhos具有更好的整体性能。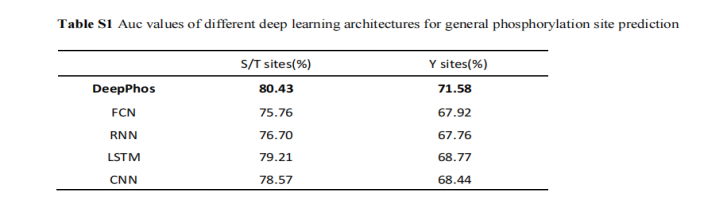
本文将DeepPhos与几种现有工具进行了比较,以使用独立的测试数据预测一般磷酸化位点。对于S / T和Y位点的一般磷酸化预测,使用几个著名的预测工具,包括NetPhos3.0,PPSP,Musite和MusiteDeep进行比较。总的来说,DeepPhos的性能比其他四个工具要好(如图2),本文又
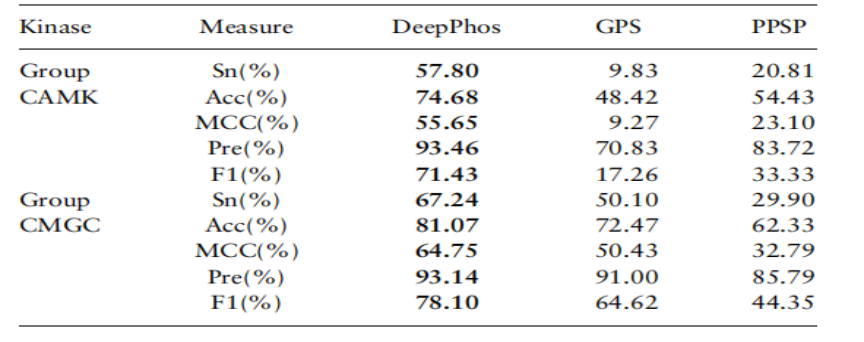
将DeepPhos与一些现有的激酶特异性预测工具进行了比较,包括基于独立测试数据的PPSP,GPS和MusiteDeep。在S / T和Y位点上选择了一些激酶组,家族,亚科和个体激酶,这些激酶的样本量最大,用于性能评估。评估结果表明,在激酶特异性磷酸化位点预测中,本架构的性能也较好(如表2)。
图2.不同预测工具的一般磷酸化S/T和Y位点预测的性能比较
表.2不同预测工具激酶特异性磷酸化位点预测的性能
五、总结
文章提出了DeepPhos,这是一种用于预测蛋白质磷酸化的新型深度学习架构。与多层卷积神经网络不同,DeepPhos由密集连接的卷积神经元网络块组成,可以捕获序列的多种表示形式,通过块内串联层和块间串联层进行最终磷酸化预测。DeepPhos还可用于激酶特异性磷酸化位点预测,不受组,家族,亚科和个体激酶水平的影响,且性能较好。








