本文介绍一篇来自浙江大学侯廷军教授课题组、中南大学曹东升教授课题组、华东理工大学李洪林教授课题组联合发表的论文。该论文提出了一种能够在分子生成过程中考虑到蛋白-配体相互作用的深度学习生成模型RELATION,该模型适用于基于靶标结构的全新药物设计。RELATION模型同时使用百万量级的分子库以及蛋白-配体集合数据对变分自编码器进行训练,在引入双向迁移学习后,隐藏层的采样能够同时兼顾生成分子的骨架片段的新颖性以及对靶标蛋白的亲和性。RELATION模型还提供了药效团约束生成以及贝叶斯优化(BO)采样等模块,可供用户定制化生成药效团匹配度更高以及对靶标的对接打分表现更好的分子。
先导化合物的发现与优化在新药研发过程中至关重要,高质量的先导化合物能够大大缩短药物探索的时间,提高成药的可能性。在先导化合物的设计过程中,要充分考虑候选分子的结构新颖性、生物活性、靶标选择性、化学可合成性、成药性和安全性等,这些性质直接影响药物开发的成败,因此先导化合物的发现一直是创新药物研发的主要瓶颈。随着计算机硬件、软件和算法的飞速发展,高通量筛选虚拟筛选和药物从头设计等计算机辅助药物设计技术开始取代传统方法,并大大缩短了先导物发现的时间和成本。
全新药物设计与虚拟筛选技术不同,不依赖已有的化学数据库,可以通过不同的生成算法对类药空间进行更加深入的探索和发掘。传统的全新药物设计方法通常将遗传算法结合到药物从头设计中,尝试通过进化策略来优化生成的化合物结构。然而,传统的药物从头设计方法无法兼顾生成分子的新颖性与理想属性。深度学习(Deep learning, DL)的引入为全新药物设计注入了新的活力。作为近期发展最快的人工智能技术,DL能够更高效地处理数据,对化合物属性深度特征的提取能力更强。鉴于深度学习对分子属性的深度特征出色的提取能力,目前已经有近百种基于深度学习的框架的全新药物设计模型被开发出来,旨在解决传统方法中生成分子的新颖性与理想属性之间的冲突。这些方法大致可以被分为四类:编码-解码器(Encoder-Decoder,Enc-Dec)、循环神经网络(Recurrent Neural Network, RNN)、生成对抗网络(Generative Adversarial Networks,GAN)和强化学习(Reinforcement Learning,RL)。
目前大部分基于DL的全新药物设计模型是以配体为中心,配体分子被表示为SMILES字符串或2D分子图。这些基于配体分子的全新药物设计的模型,在经过训练后确实能够生成大量有效且新颖的化合物,但是这些基于配体二维信息的表征会忽略分子在药物设计任务中一些非常重要的属性,比如药物分子的三维立体构象以及与蛋白之间的结合构象。本文介绍的RELATION模型是一个使用了变分自编码器框架的生成模型,在双向迁移学习的作用下,模型能够生成大量结构有效、结构新颖并且对蛋白具有一定亲和力的化合物。在药效团约束和BO采样的作用下,RELATION将会更加适用于基于靶点结构的全新药物设计任务。
RELATION模型的训练使用了源域和目标域两种数据集。源域的百万数量级的小分子化合物来源于ZINC数据库。目标域则使用AKT1以及CDK2两个靶点的数据集,407个AKT1抑制剂和1017个CDK2抑制剂搜集于BindingDB和ChEMBL数据库,然后将两个靶点的抑制剂对接到靶标蛋白,只保留配体周围5 Å的原子作为蛋白配体复合物数据集。随后将源域数据集和目标域数据集放入7.57.57.5 Å3的网格中,并将源域数据集和目标域数据集的质心与立方体框的质心对齐,重原子的位置以1 Å作为分辨率,每个原子由19个物理化学性质描述。最后,源域数据集和目标域数据集中的每个分子都由一个由其坐标特征向量定义的四维张量表示。
RELATION框架由两个部分组成:(1)3D编码器,使用了3D-CNN的结构,包括私有编码器和共享编码器。附带SMILES标签的训练源域数据以及目标域数据转换成4D张量后,分别作为私有编码器和共享编码器的输入。所有的编码器具有相同的架构,均具有8层,第一层包含64个过滤器,然后在奇数层上加倍,最后一层学习512个过滤器。每一个偶数层后面都有一个额外的池化层,核数、步长和填充为2,用于执行下采样。利用ReLU激活函数对3D-CNN模型进行训练,并使用两个输出为512维的全连接层得到μ和σ,对其重参数化后,生成一个的1024维嵌入向量;(2)解码器,解码器的结构是caption-LSTM,可以将隐藏层内的高维向量转化为SMILE分子式,caption-LSTM由三层组成,其词汇量输入大小为39,隐藏大小为1024。
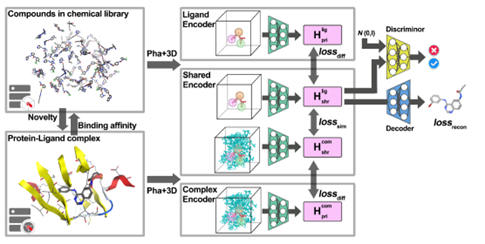
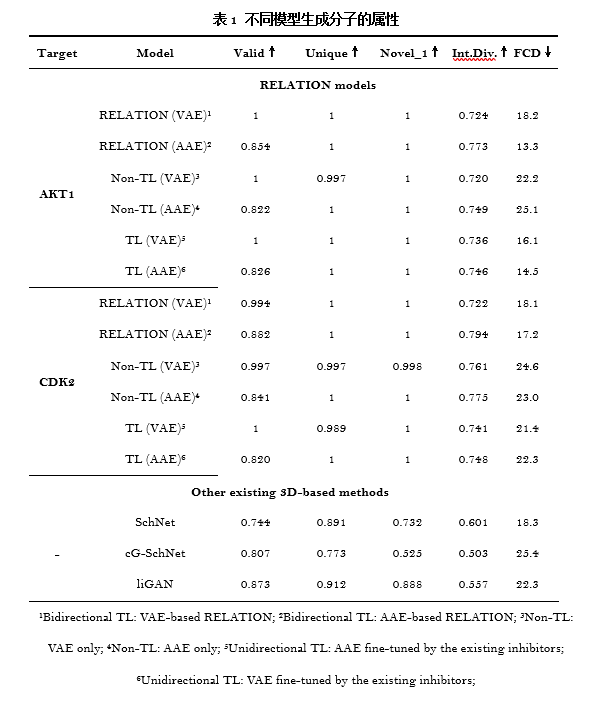
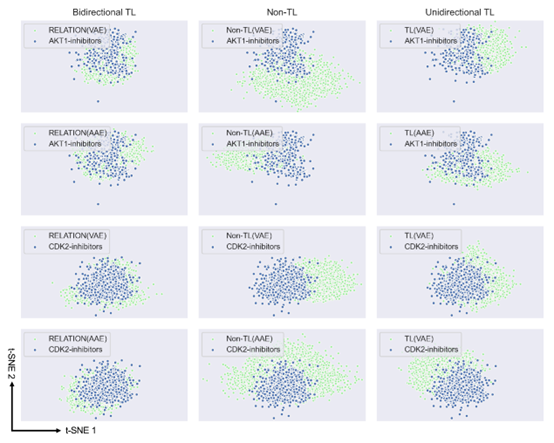
表1中的计算结果显示,RELATION模型生成的分子的有效性、独一性、新颖性以及生成的分子的多样性均优于其他3D生成模型。随着双向迁移学习的引入,可以发现双向迁移学习的RELATION (AAE)和RELATION (VAE)模型的有效度、独一性以及多样性均高于其他模型。图1的结果显示非迁移学习框架生成的分子的分布与抑制剂完全不同,当使用单向迁移学习框架对模型进行再训练时,生成的分子分布与现有抑制剂分布的重叠明显增加。而使用双向迁移学习RELATION框架后,模型产生的分子的化学空间分布与抑制剂完全重合,表明所生成的分子和现有抑制剂涵盖了类似的化学空间,并且具有相似的属性。图2则展示了不同模型生成的分子与AKT1和CDK2抑制剂的化学空间分布。图2的结果也与表1中数FCD数据一致。这些结果均表明,RELATION模型生成的分子不仅能够保证有效性、新颖性以及多样性,也能够保证和已有抑制剂的属性相似性。
RELATION模型的药效团约束是通过CVAE框架实现,将药效团特征匹配度作为CVAE的特征引入到RELATION的训练中。不同模型生成分子的药效团分数分布如图3所示。对于AKT1和CDK2,基于药效团的RELATION模型产生的分子比原始RELATION模型产生的分子有更高的药效团分数。这表明,通过将药效团特征引入RELATION,生成的分子可以增强与预设药效团模型间的匹配性。
作者还在RELATION框架中引入了BO的采样。如图3所示,在RELATION框架中引入BO采样后生成的分子的药效团分数都得到了提高,其中基于对接打分的BO的采样性能略好于基于QSAR打分的BO。此外,基于对接打分的BO采样产生分子的对接分数较原始RELATION模型生成分子的对接分数有明显提高,但基于QSAR打分生成的分子的对接打分变化不大。

为了进一步研究基于BO采样的RELATION模型的性能,作者将不同模型生成的有效分子与AKT1抑制剂再次进行了T-SNE分析。如图4所示,RELATION和RELATIONpha模型不能有效地探索AKT1抑制剂的化学空间(红圈中标记的点)。随着通BO-采样方式的引入,生成的分子在化学空间中的分布比原始RELATION更加分散,说明生成的分子与AKT1抑制剂的化学空间更为相似。此外,根据点的颜色梯度,使用BO采样的RELATION模型生成的分子比原始RELATION模型生成的分子的对接得分更优。
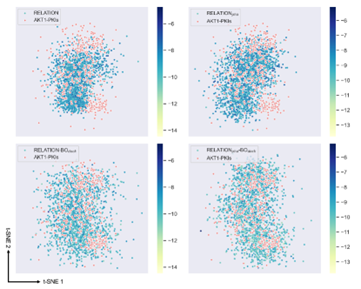
图4 RELATION模型使用不同的采样方式生成分子的化学空间分布作者在图5中展示了不同RELATION模型生成的一些分子的示例。如图5所示,引入BO采样后,RELATION和RELATIONpha均能生成对接分数较好的分子,但基于BO采样的RELATION模型生成的分子药效团匹配分数较高,并产生了更理想的药效团特征。
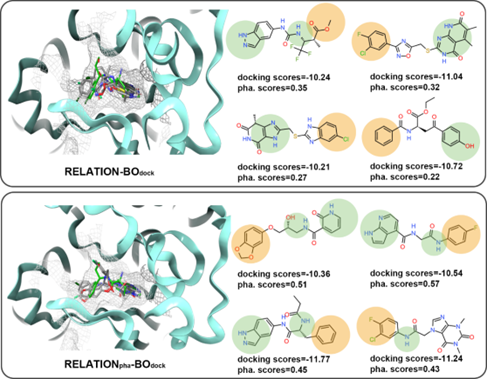
图5 使用RELATION模型的设计AKT1抑制剂实例
在AKT1与CDK2的抑制剂全新设计的任务中,RELATION模型既能生成结构新颖且多样性高的分子,并且能够保证生成的分子对靶标具有一定的亲和性。随着基于对接打分的BO采样以及药效团约束模块用于RELATION模型,RELATION模型能够使得生成的分子同时具有更好的药效团匹配和对接表现。这些结果表明,RELATION模型是一种极具竞争力的深度学习全新药物设计模型。
RELATION: A Deep Generative Model for Structure-based De Novo Drug Design, Journal of Medicinal Chemistry, 2022.
https://doi.org/10.1021/acs.jmedchem.2c00732








