随着医疗技术的进步,人类的健康水平获得了极大提升。但面对给人类健康带来巨大威胁的癌症,尚没有十分有效的治疗手段。据世界卫生组织统计报告,2012年全世界估计约1410万人被检测出癌症,并造成820万人死亡(相当于全年死亡人数的14.6%)。男性最常见的癌症包括肺癌(LUAD)、前列腺癌(PRAD)、结直肠癌(COREAD)以及胃癌(Stomach cancer);女性常见的乳癌(thoracic cancer)、结直肠癌(COREAD)、肺癌(LUAD)和宫颈癌(UCEC);儿童是白血病(ALL)和脑瘤(Brain tumour)最为常见[1]。
在整个生命过程中,细胞DNA受损后导致基因突变,并引发一系列机体反应,最终引起癌症的发生。人类对癌症的研究已经进入基因组时代,科学家们将癌症相关基因进行了整理和标记[2,3]。这些癌症相关基因又被分为驱动癌基因(driver)和从动癌基因(passenger),对于基因间的关系如何,对肿瘤发展的影响水平大小以及这些基因在不同肿瘤之间的不同突变频率具有怎样的意义等问题,在很大程度上仍然是个未解决的问题。已有研究表明,例如肿瘤蛋白TP53,表皮生长因子受体EGFP和β连环蛋白CTNNB1突变与大多数癌症的发展有关。同时,绝大多数癌基因突变发生在中性突变之后,对特定三核苷酸变化具有明显偏好。
近日,巴塞罗那生物医学研究所研究团队通过模拟中性突变,用机器合成一组推测的从动癌基因突变,以此来模拟在人体内癌症相关基因的多米诺骨牌式突变。研究团队基于机器学习的方法开发出一种算法“boostDM”(图1),用于评估给定基因所有可能的诱变,并评估它们对癌症发展影响的可能性。该算法可帮助临床医生对于病人的癌症发展做出更准确的解释。该研究成果发表在Nature上,文章题为“In silico saturation mutagenesis of cancer genes”。
文章的通讯作者López-Bigas表示:“该算法可模拟特定类型癌症的每个基因中的每个可能突变,并指出哪些是癌症过程中的关键。这些信息有助于我们在分子水平了解肿瘤是如何引起的,促进癌症患者进行最适合治疗的医疗决策。”
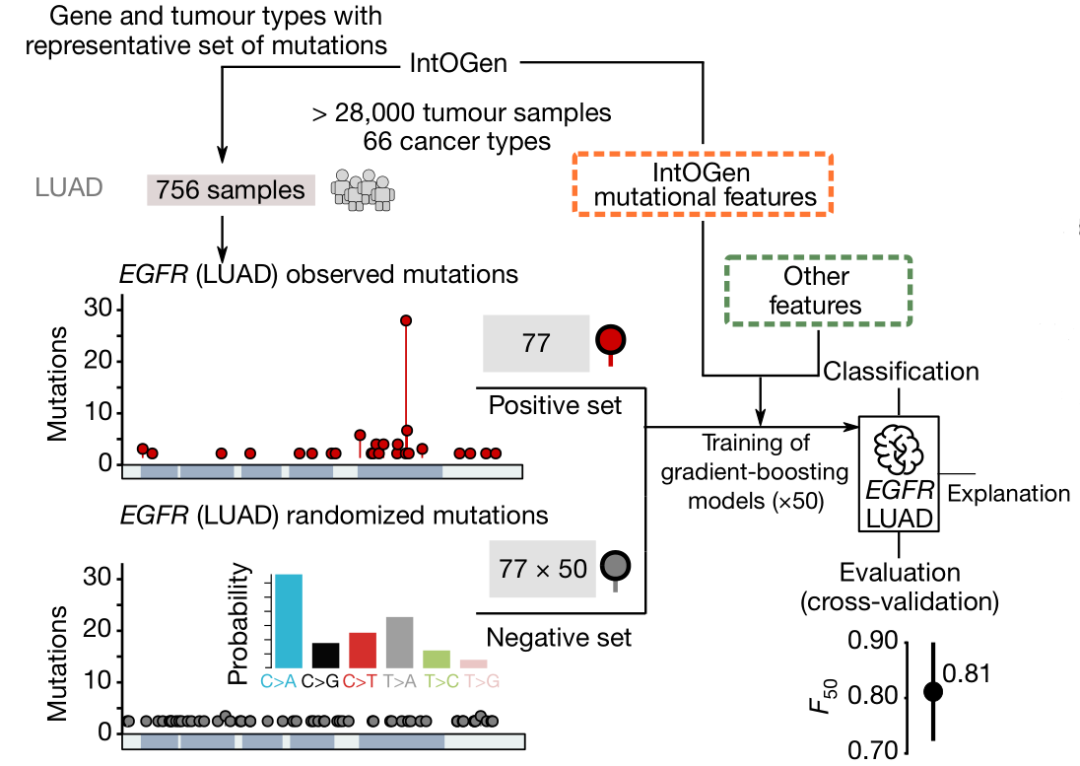
图1.用肺癌(LUAD)样本对机器学习模型boostDM的训练和交叉验证。来源:Nature[4]研究人员从公共数据库中收集了来自66种癌症类型约28,000个肿瘤样本数据,确定了568个癌症基因突变。研究发现,与随机突变相比,在样本中发现的驱动癌症生长和发展的突变数量更多。
研究团队通过交叉验证检测了boostDM模型的性能。从图2中可以看出,boostDM模型比其他算法更加精准(precision)和更高的发现能力(recall)。boostDM获得的数据和实验数据具有较高的吻合度。
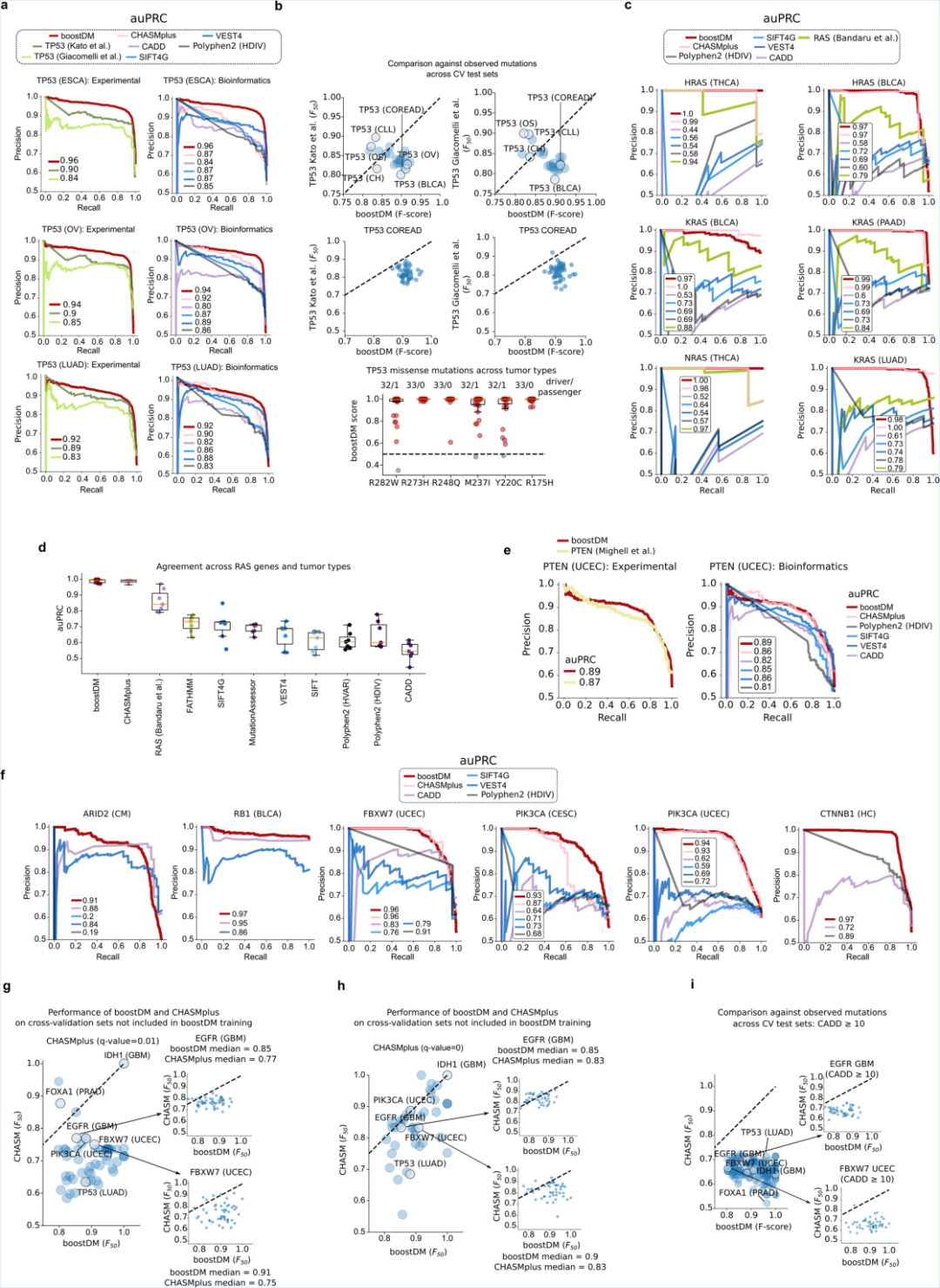
图2. boostDM模型优于其他算法。来源:Nature[4]研究人员在肺癌与胶质母细胞瘤中鉴定驱动癌基因和从动癌基因。(图3)分析发现,两种癌细胞中驱动癌基因的分布不一致,反映了肿瘤发生的不同机制。图3D中SHAP值越大的基因表示该基因对癌症的发展产生的驱动效应越强,越小则表示其对驱动因素负面作用回馈。
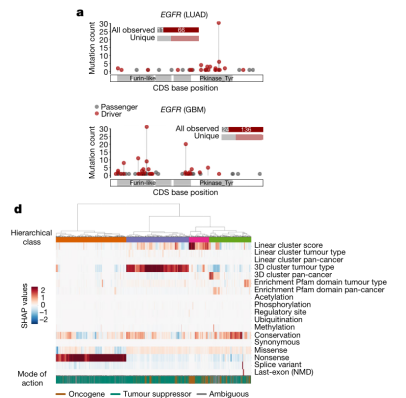
研究团队将185个癌症基因-组织模型应用于基因序列中所有可能的核苷酸变化进行实验,以绘制全面的不同蛋白质区域具有跨肿瘤类型驱动突变的可能性的图谱(图4)。
图4.TP53和CTNNB1的驱动突变图谱,EGFR在三种肿瘤组织中的突变图谱。来源:Nature[4]该突变图谱揭示了许多从未在肿瘤中观察到的潜在驱动突变。为探索突变概率对驱动突变的影响,研究人员计算了不同肿瘤类型癌症基因中所有潜在驱动突变的发生概率。结果显示,大多数癌症基因表现出强烈的突变概率偏倚,肿瘤抑制基因通常表现出比癌症基因明显更强的偏倚。这意味着,对于大多数癌症基因,潜在驱动突变的发生概率会影响它们中的哪些基因会被检测到。
正如文章作者所说:“该研究结果报表明,应用受进化生物学启发的机器学习方法来构建高质量模型是可行的。这些模型从人类肿瘤中检测到的突变中学习,以识别癌症基因中的驱动突变。”
到目前为止,研究团队已经利用BoostDM制作了185个模型来识别不同类型癌症的突变。例如,确定了导致肺癌肿瘤的EGFR基因中所有可能的突变,并在另一个模型中确定了胶质母细胞瘤脑肿瘤。研究人员计划继续开发和改进 BoostDM,随着越来越多不同肿瘤测序数据的加入,应该会变得更加准确。
参考资料:
[1]. World Cancer Report 2014. World Health Organization. 2014: Chapter 1.1. ISBN 9283204298.
[2]. Sondka, Z., Bamford, S., Cole, C. G., Ward, S. A., Dunham, I., & Forbes, S. A. (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nature Reviews Cancer, 18(11), 696-705.
[3]. Martínez-Jiménez, F., Muiños, F., Sentís, I., Deu-Pons, J., Reyes-Salazar, I., Arnedo-Pac, C., ... & Lopez-Bigas, N. (2020). A compendium of mutational cancer driver genes. Nature Reviews Cancer, 20(10), 555-572.
[4]. Muiños, F., Martinez-Jimenez, F., Pich, O., Gonzalez-Perez, A., & Lopez-Bigas, N. (2021). In silico saturation mutagenesis of cancer genes. Nature, 1-5.
· END ·
 热文推荐
热文推荐









