Dr. X 是由晶泰科技博士团发起的专业知识分享栏目,旨在向生物医药行业传递和分享全球 AI 药物研发的前沿技术与研发动态,促进广大药物研发从业者对 AI 药物研发的认知,推动 AI 等前沿技术在药物研发领域的应用。
现代药物研发是一项漫长且复杂的工作流程,有统计数据显示,每个成功上市的化学药物背后,平均经历了十万个化合物筛选和数百项临床前实验【1】。
分子结构设计是合理药物设计的基础,也是获得全新药物分子的起点,肩负着创新型分子成药性、专利空间、提高可合成性等诸多使命,其核心问题是从合成可及的化学空间中获得满足特定活性和成药性的候选分子。深度学习指导下的全新分子设计作为一种不同于虚拟筛选的化学空间探索方式,通过运用神经网络,在给定的分子库中学习化学结构特征和化学结构设计的规则,并将这些规则运用到基本分子构建单元的堆砌、符合构效关系的分子片段或药效团链接等,指导药物分子设计与生成。由于所使用的数据集未经标记分类,因此深度生成模型可大致归属于非监督学习,这意味着生成模型具有较广的探索空间。
本质而言,基于深度学习的全新分子设计方法着重关注于如何估算分子结构的分布 𝑝(𝐱) 。全新分子生成则可以视为于从𝑝(𝐱)中抽取样本𝐱 ~ 𝑝(𝐱);相应地,此过程需要使用参数化的概率分布𝑝𝜃(x) 来逼进分布 𝑝(x)。理论上,当我们了解了这个分布规律,就可以获知整个数据集的信息,也就是满足要求的全部化学结构。结合打分函数对生成过程进行指导,就可获得该空间内符合要求的新分子结构。
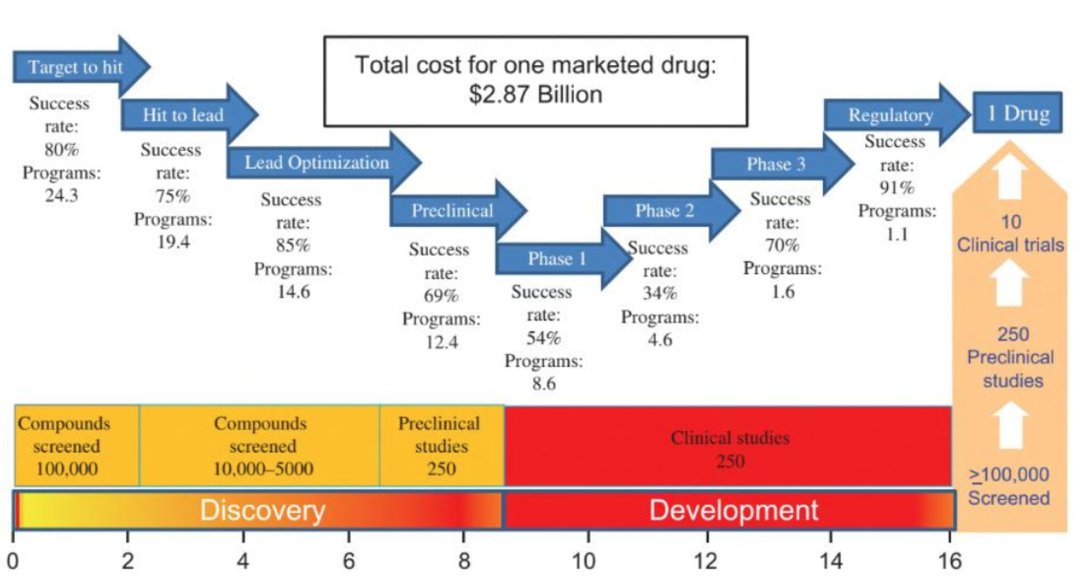
每个成功上市的化学药物都以海量的设计-合成-测试-分析的循环迭代实验结果支撑,耗时漫长且成本高昂。
常用的分子生成模型构架主要包括循环神经网络(Recurrent neural network, RNN),变分自编码器(Variational autoencoder, VAE),生成对抗神经网络(Generative adversarial network, GAN)和流模型(Generative glow-based model)等。每种模型因其架构特征都存在各自的优势和局限。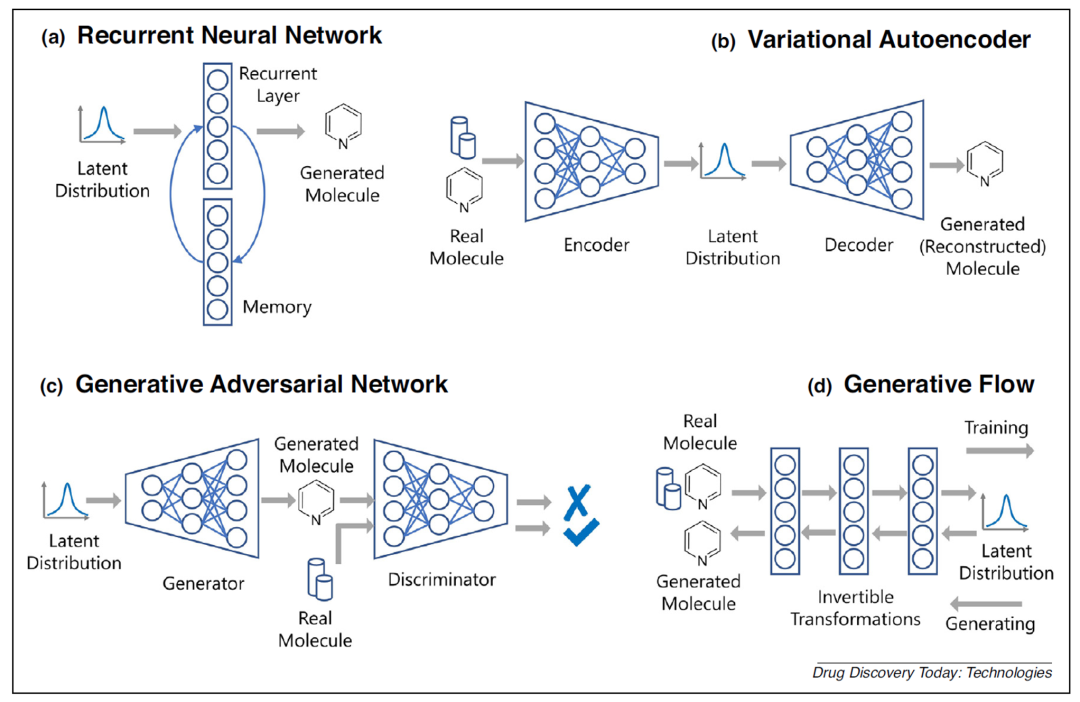
• RNN 网络的结构可概括为将前一步骤的输出用作下一步骤的输入,这种“记忆”效应有助于决定对新输出的阐述方式,同时兼顾保持原始输入的特性。
• VAE 模型已经被成功用于图像、文本等多种类型的数据生成任务中,其特色在于存在隐变量空间,在训练VAE时模型倾向于在隐藏变量中存储最有价值的有压缩意义的信息,可用于许多后续学习任务或移植到其他类型的模型之中。
• GAN 的数据逼真度较高,但网络训练的本质不再是优化问题,而是寻求纳什均衡,不仅较难训练,也容易遭遇模型坍塌。考虑到 GAN 同时具备生成和判别的功能模块,因此 GAN 模型结构在对化合物分子向特定性质优化时具备潜力,比如明确成药性、水溶性、抑制活性、指定芳香环数量等性质要求【3】。
掌握了基础算法工具接下来就要展开应用。首先需要解决的问题就是如何把人看来简单的化学结构,转化成能让计算机识别的信息。只有满足这个条件,才好利用底层的成熟算法去训练模型。目前多种的化合物分子表征方式被用于计算机对化学分子结构及其特征的识别,由此也衍生出多个层次的分子生成方式。
简化分子线性输入规范(Simplified Molecular Input Line Entry Specification, SMILES)是最常见的化学分子描述符,将化学结构以字符串表示,构建类似于语言结构。RNN 模型见长于对序列属性的信息处理,比如自然语言识别、乐曲生成等。二者的结合令 RNN 模型通过训练集学习到 SMILES 序列每个位置出现特定原子字符的几率,进而获得化学结构和化学空间的分布规律,以指导对已有字符串进行字符替换或产生全新字符串的工作,这就是一维生成模型的工作方式。一维分子生成模型也可通过 VAE 网络实现,通过控制隐藏变量来操控分子的结构,且发现 VAE 训练得到的隐藏表征与结构存在较高的关联性【4】。
基于 SMILES 的生成模型在很多生成任务中已经有过很好的应用和表现能力。然而 SMILES 对分子结构的细微变化的描述并不稳健;另外,此类模型所生成的 SMILES 字符串需要极为严格的语法限制才能被解码为有效的分子,这增加了模型学习的成本。可见,亟待建立更加稳健的分子生成模型。
鉴于一维生成模型生成线性字符串的分子形式具有的局限性,促使研究向更具表现力的“分子图”方法推进。分子图是一种标记图,由对应于原子的点和对应于化学键的边组合而成,并通过一定的连接关系连接到一个集合中【5】。图生成模型在分子图生成领域有着特有的优势,并着重于解决以下两个问题:(1)设计能够直接处理图数据结构的神经网络架构;(2)设计分子图的生成过程。
基于片段(Fragment-based)或砌块(Building blocks)等构建单元进行拼接生成新骨架的方式是二维分子生成模型的基本思路。可采用 VAE 编码器将分子编码成若干亚结构(构建单元)和亚结构间结(Node)的连接方式,通过训练集学习构建单元的特性和出现频率,和结的拼接规则,最后通过解码器将新拼装出的亚结构组合进行解码,就获得了全新的分子图。
值得一提的是,尽管用于堆砌连接构建新分子的片段种类有限,事实上,即时在使用几个固定构建单元进行拼装时,深度学习网络产生出的结差异巨大,令最终产生的分子仍具有较高创新性【6】。
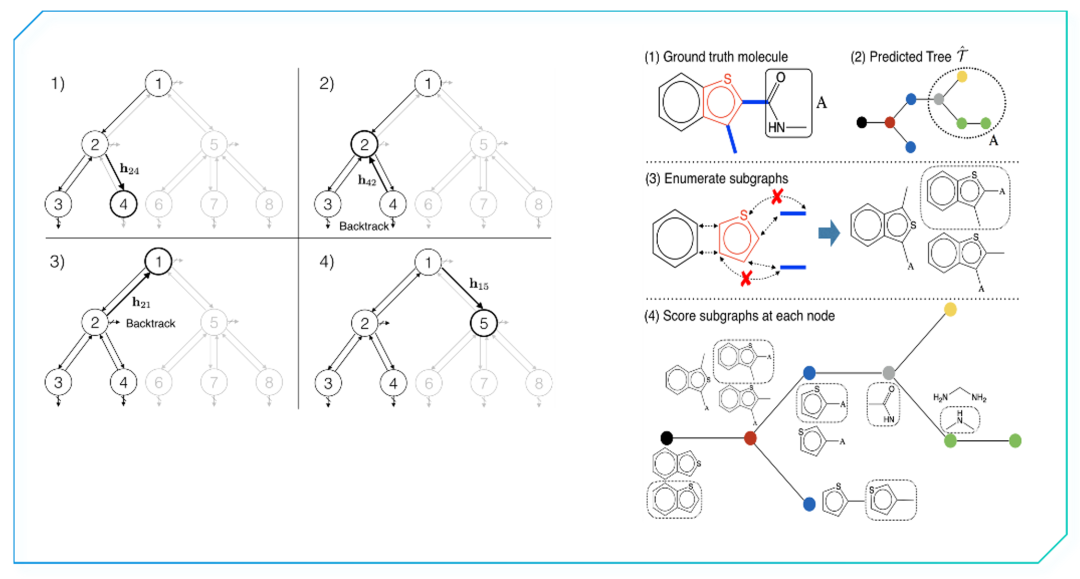
在生成的每一步都对树结构 (Junction tree) 的分支结构和性质进行判断的同时,还将信息返还至上一层次的节点(左)。对每个结位置连结上的堆砌单元进行评分(右)。前述两种分子生成模式均着眼于对配体结构特征的提取。综合考虑靶点口袋和化学分子的三维构象进行分子设计是一种基于形状(shape-based)的三维分子生成模型。生成对抗网络的运用可以生成与结合口袋互补的配体三维结构。一方面,将结合口袋以图的表征方式输入并编码为隐藏向量;另一方面,训练网络运用隐藏向量信息生成满足口袋结构的配体形状【7】。
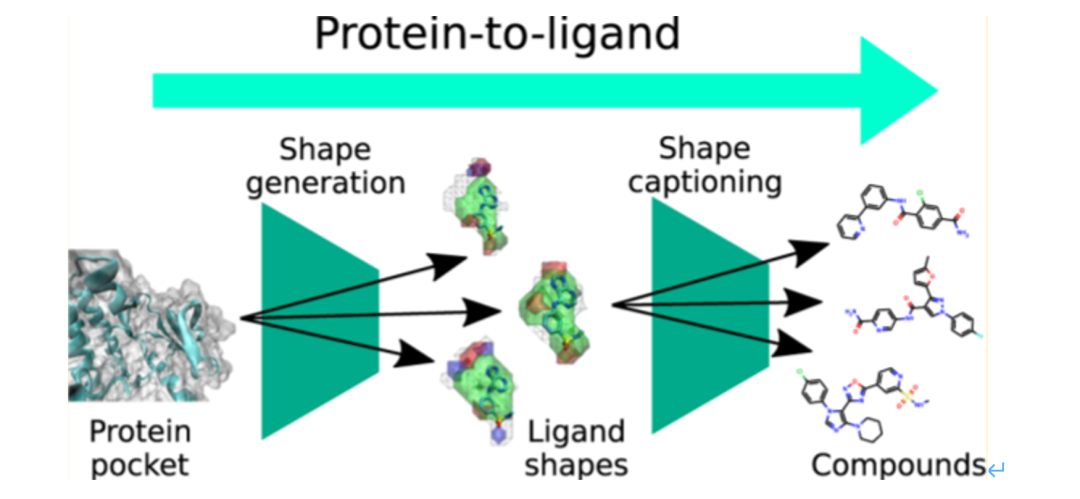
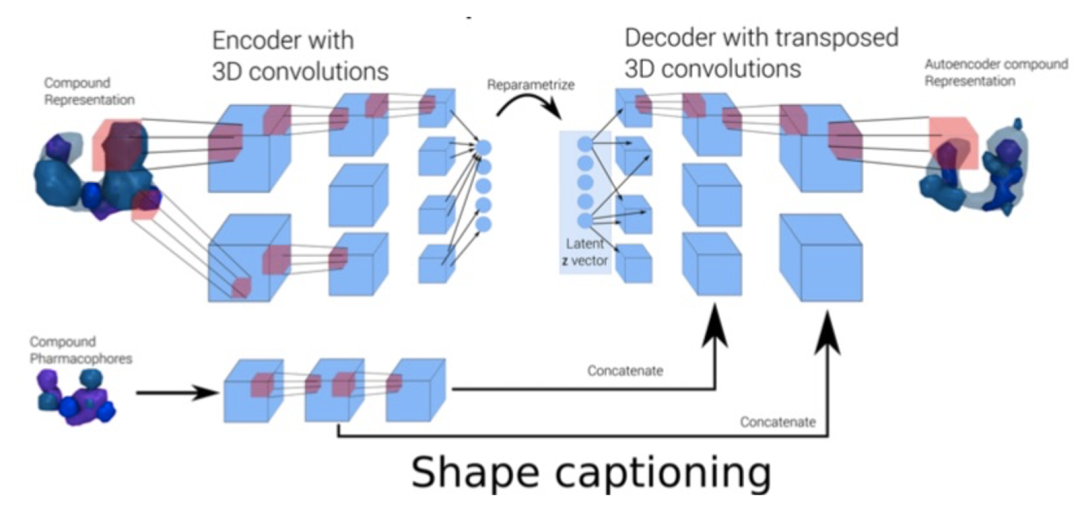
也有研究报道结合图卷积模型和变分自编码器构建了分子三维生成模型。首先对分子的构象进行格点像素的表征并编码成为 3D 药效团结构,经过 VAE 的扰动之后,再解码成新的 3D 药效团结构,就达到“从药效团到新药效团”的生成目的【8】。
根据药物设计需求进行全新分子设计需要解决的核心问题是如何在已有的化合物设计规则之中探索新的化学空间。基于深度学习的生成模型将这一问题化归为对条件概率密度的估计。深度分子生成模型拥有从大量数据中学习的能力,以及超越化学直觉的从头药物设计的潜力,已被广泛应用于许多重要的分子设计项目当中。尽管目前模型的可靠性大多通过虚拟的药物设计任务得到测试,生成的化合物只有在合成之后,其成药潜力才能被客观测试。但基于 AI 的性质优化与预测将待合成测试的分子数收敛至数十个,这是传统药物研发手段所不能实现的。自动化实验技术等的发展也必将推动数据的快速获取。在不久的未来,我们或许就可以看到 AI 分子生成算法与自动化技术完全地结合,实现较少(甚至没有)专家干预的药物设计流程。
Dr. X 科学家们在这些初级应用的基础上针对特定场景做了二次的开发,开发出了更多满足实际应用需求的定向优化模型,比如 Random(无序、随机的生成),SIMILAR(相似生成),SCAFFOLD(固定母核的生成),SCAFFHOP(骨架跃迁),REPLACE(侧链替换),PHARM(药效团),POCKET(基于小分子形状或蛋白口袋)等。
优化分子的话题将在下期开展,敬请期待。
参考文献
【1】B. E. Blass, Basic principles in drug discovery and development, ISBN: 978-0-12-411508-8 (2015)
【2】Xia, X. et al. Drug Discov Today Technol 2019, 32, 45–53.
【3】Maziarka L. et. al. J Cheminform. 12(1):1–18 (2020).
【4】Blaschke, T. et. al. Mol. Inform. 2018, 37, 1700123.
【5】Biggs, N. et. al., Graph Theory, 1736-1936 (1986).
【6】Roughley. S. et. al. Top. Curr. Chem. (2011)
【7】M. Skalic, et. al. Mol. Pharmaceutics, 16, 4248-4291 (2019)
【8】M. Skalic, et. al. J. Chem. Inf. Model, 59, 1205-1214 (2019)
晶泰科技(XtalPi)是一家以数字化和智能化驱动的人工智能(AI)药物研发科技公司,基于量子物理、量子化学、人工智能与云计算技术,为全球创新药企提供智能化药物研发服务。我们希望通过提高药物研发关键环节的效率与成功率、降低研发成本,助力合作伙伴为患者带来更多优质的药物。晶泰科技创立于麻省理工学院(MIT)校园,核心团队由来自学术界、IT 互联网界及医药产业界的优秀人才组成。公司总部位于深圳,并在北京、上海、波士顿设有分部,成立至今,晶泰科技已经成功为来自美国、欧洲、中国、日本的近百家先锋药企提供了药物研发服务。
 点击这里,更多了解晶泰科技!
点击这里,更多了解晶泰科技!










