今天给大家介绍山东大学魏乐义教授等人在Bioinformatics期刊上发表的文章“Predicting protein-peptide binding residues via interpretable deep learning”。识别蛋白质与多肽的结合位点对于了解蛋白质功能机制和探索药物发现至关重要。尽管前人已经提出了许多相关的计算方法来解决这一问题,但这些方法大都高度依赖第三方工具或信息进行特征提取与设计,容易导致计算效率低下、预测性能不高。为了解决这一问题,作者提出了PepBCL,这是一种新的基于BERT的对比学习框架,仅基于蛋白质序列预测蛋白质-多肽结合位点。PepBCL是一个独立于特征设计的端到端的预测模型,在基准数据集上显著优于许多SOTA方法。此外,作者团队还探讨了PepBCL中注意力机制对于蛋白质结合区域中结合位点周围残基序列特征的挖掘能力,从而对模型如何预测结合位点进行了一定的解释。最后,为了方便研究人员使用,作者团队还搭建了一个在线预测平台作为所提出的PepBCL的实现,其服务可以访问如下网址:https://server.wei-group.net/PepBCL/。
蛋白质-多肽相互作用是重要的蛋白质相互作用之一,在许多基本细胞过程中起着至关重要的作用,例如DNA修复、复制、基因表达和代谢等。研究还发现,蛋白质相互作用涉及一些异常的细胞行为,这些行为会诱发多种疾病,其中约 40%的相互作用是由相对较小的多肽介导的。因此,识别参与蛋白质-多肽相互作用的结合位点对于理解蛋白质功能和药物发现都是必要的。目前现有的计算方法或多或少存在如下问题:1)在缺乏相关的肽结合蛋白结构时,基于蛋白质结构的结合位点预测方法则无法进行预测;2)目前大多数方法都依赖于第三方工具包来提取预测特征或者进行手工特征提取来预测结合位点,但这会导致预测效率的大大降低;3)蛋白质-多肽结合位点数据集是一个极度不平衡的数据集,关于如何提升在不平衡数据集上的位点预测性能,之前的工作很少涉及。基于此,作者团队提出了PepBCL,该模型可以仅输入原始蛋白质序列进行预测蛋白质-多肽结合位点,不需要借助其他工具和手工特征设计,而且通过提出的对比学习框架,该模型可以有效地提高不平衡数据集上的预测性能。
PepBCL包括1)序列嵌入模块、2)基于BERT的编码模块、3)对比学习模块和4)输出模块四部分,如图1所示。
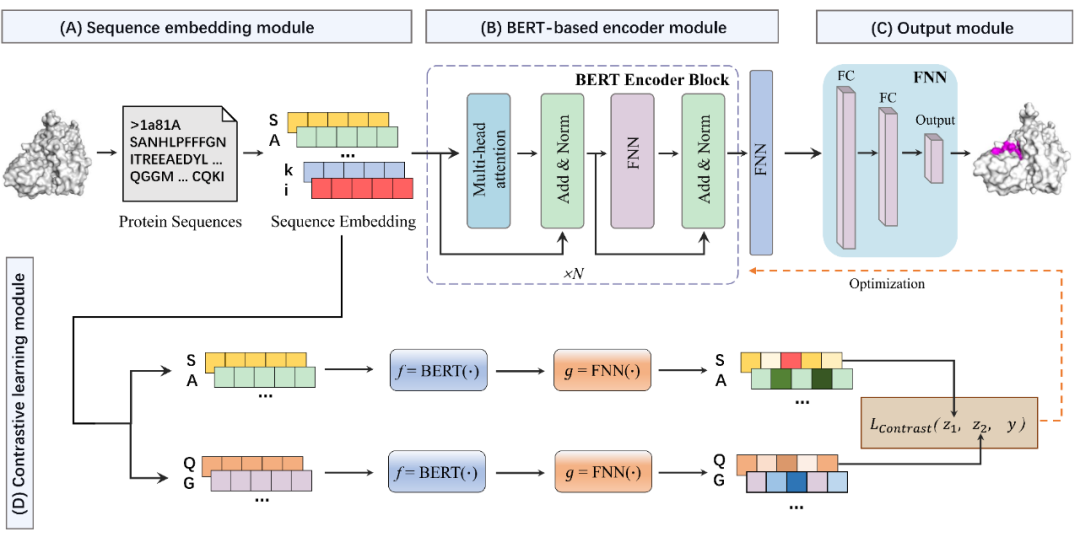
图1. PepBCL模型的网络架构
在该模块中,原始蛋白质序列将被编码为一个嵌入矩阵;具体方法为:原始蛋白质序列中的每个氨基酸字母首先被大写,并根据定义的词典翻译成数字序列,其中序列中的每个氨基酸都看作是句子中的一个词,并映射到一个数字值。通过这种方式,原始蛋白质序列被编码为数字值向量。然后,这些数值序列通过查找表得到相应的嵌入向量,该查找表是一个嵌入层,与基于BERT的编码器模块同时在大量蛋白质序列中进行了预训练。最后,原始蛋白质序列中的每个氨基酸都得到了相应的嵌入向量,从而使得整个蛋白质序列也被编码为嵌入矩阵。
BERT模型的基本单元是由多头注意力机制、前馈网络和残差连接技术组成的编码器块。多头注意力机制由许多独立的自注意力模块组成,用于学习蛋白质序列的多角度上下文表示。自注意力机制描述如下:
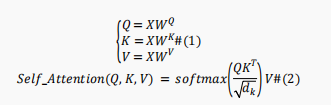
多头注意力机制基于上述的自注意力机制,可表述为如下:
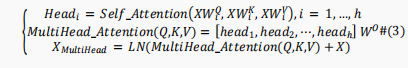
加入前馈网络(FNN)以通过激活函数提取更好的表示,其数学描述如下:
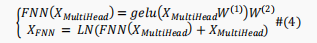
由于BERT模型有许多编码器块,因此BERT的最终编码过程可以表示为如下:
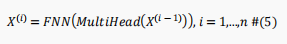
BERT模型编码后,将得到最后一个编码器块的输出,这时候维数仍然很高。因此,为了避免维度的冗余,如下使用全连接神经网络来更好地提取输入序列中氨基酸的表示,同时降低维度:
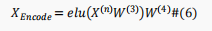
在这里,作者提出了一种基于有监督数据的新颖的对比学习模块,使得相同类别输入的表示映射到表示空间中相近的点,而不同的类别输入则映射到远处。具体来说,为了使相同类别样本具有相似表示而不同类别的样本具有不同的表示,作者构建了对比损失作为模型针对批量数据的损失函数。对于一个batch中的一对位点表示,损失定义如下:
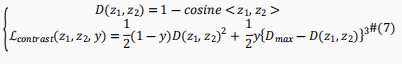
值得注意的是,作者给不同类别位点对一个更高的权重3,使模型间接地更多关注少数类。
由前面的模块从原始蛋白质序列x中生成的位点表示向量z被送入全连接网络,将特征向量转换为位点级别的类别输出,即,
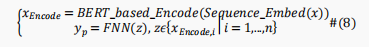
这里使用交叉熵损失函数来训练输出模块以提高预测性能,即,
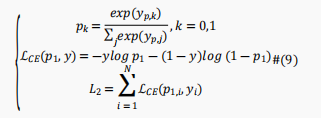
为了避免交叉熵损失的反向传播干扰表示学习模块和由于深度模型BERT引起的梯度消失问题,表示学习部分的优化和预测部分被分离开来。具体来说,作者在训练输出模块时冻结基于BERT的编码器模块中的参数。最终模型的损失函数可描述为如下:
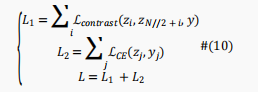
作者首先对PepBCL和现有方法在独立测试数据集上进行了性能比较,如表1所示,可以看出就AUC和MCC而言,PepBCL好于所有基于序列的模型和大部分基于结构的模型。值得一提的是由于PepNN-Struct方法在预测结合位点时不仅使用了蛋白质结构数据而且还使用了来自多肽的信息,因此该方法的AUC取得了最高效果,而PepBCL仅使用了蛋白质序列作为输入信息。
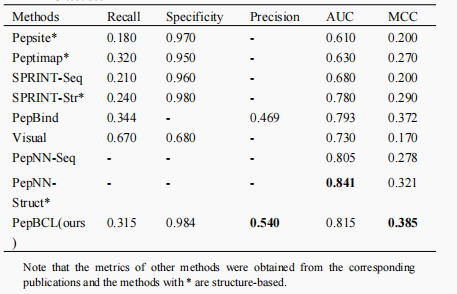
表1. 与现有方法进行性能对比
为了验证提出的对比学习模块能够帮助模型提取到更加高质量的特征,作者进行了消融实验,如表2所示,
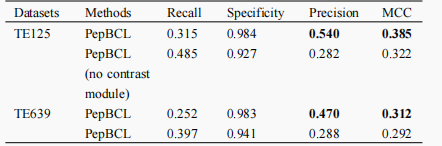
表2. 测试模型消融对比学习框架的性能对比
作者首先在数据集中随机挑选了一条蛋白质(PDBID:2wh6A),然后可视化了PepBCL模型在其三个结合区域的注意力图如图2中Attention maps部分,作者发现模型注意力机制对R、V和T三个残基关注度最高,因此为了进一步研究模型所能挖掘到的信息,作者又通过分析结合位点数据集获得了R、V和T三种氨基酸在结合时周围氨基酸的分布偏好并可视化在图2的Motif analysis部分,通过对比后发现1号和3号结合区域中的R和T残基周围氨基酸的分布与从数据集分析得到的R和T结合时周围氨基酸的偏好分布十分相似,因此可以推得PepBCL模型能够学习到结合残基与周围氨基酸的潜在关联;而在2号结合区域中,可以看到V周围的分布与从数据集分析得到的V结合时周围氨基酸的偏好分布就不那么相似,但是通过V也是结合残基看,模型确实关注了更可能结合的残基,作者在此推测模型不仅可以通过分析数据集获得的结合序列模式来做出预测,而且还可以挖掘出新的结合序列模式从而做出预测。
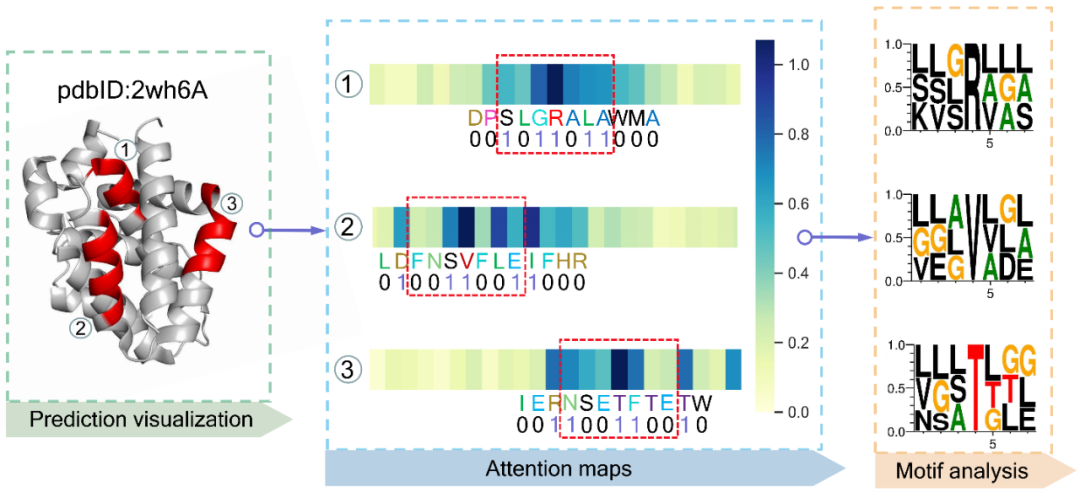
图2. PepBCL对结合区域的注意力分数的可视化
在本文中,作者团队设计并开发了一个基于BERT与对比学习的框架取名为PepBCL,用于仅使用蛋白质序列来预测蛋白质-多肽结合位点。基准数据集上的实验表明,所提出的PepBCL在所有指标方面都优于现有的基于序列的方法,在大多数指标方面都优于基于结构的方法。而且在本文中,作者还探究了模型的注意力机制在蛋白质序列分析中到底可以带给我们什么样的启发,以有助于更多的研究者加以关注和研究。为方便对此感兴趣的研究者的使用与方法比较,作者团队还开发了一个在线预测平台作为所提出的PepBCL的实现,其服务可以访问如下网址:https://server.wei-group.net/PepBCL/。
论文网址
https://doi.org/10.1093/bioinformatics/btac352
在线服务网站
https://server.wei-group.net/PepBCL/








