作者 | 杨千立
审稿 | 陈梓豪
指导 | 闵小平(厦门大学)
这次为大家分享的是来自nature communications上的一篇题为《Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space》的文章,来自密歇根大学生物界面研究所、生物医学工程系、化学工程系项目组的Peter M.Tessier团队。
治疗性抗体的开发需要高亲和力分子的选择以及其他类药物的生物物理特性,多种抗体特性的联合优化仍然是一个困难和耗时的过程,严重阻碍药物开发。在这篇文章中,作者团队提出了开发了一种简便的机器学习模型,对治疗性抗体的亲和力和特异性进行多目标优化。结果表明,机器学习模型的力量极大地扩展了对新型抗体序列空间的探索,并加速了高效的药物类抗体的开发。
抗体疗法被用于治疗人类疾病,从癌症和自身免疫性疾病到过敏和神经退行性疾病。抗体疗法的成功归功于它们的分子特性,包括它们的高亲和力、长半衰期和出色的生物物理特性。然而,从免疫接种或体外文库中选出的候选抗体通常具有广泛的生物物理特性。在许多情况下,具有最高生物活性的候选抗体表现出一种或多种阻碍生产、配制和递送的不良生物物理特性,这通常是在开发过程的后期发现的,并且可能会损害其他候选抗体的治疗潜力。因此,在开发的早期阶段,需要抗体工程方法来改善其生物物理特性,同时保持高亲和力和生物活性。不幸的是,改善给定的次优抗体特性,如特异性或溶解度,会导致其他特性(如亲和力)的缺陷。因此,迫切需要一种简单而可靠的方法来预测CDR突变,以最少的实验来共同优化抗体亲和力和各种生物物理特性。
作者团队在该项工作中,以共同优化临床阶段抗体(emibetuzumab)的亲和力和特异性(非特异性结合)特征开发预测模型,该模型可以从大量但采样稀疏的抗体CDR库中学习,并预测未在原始库中采样的新CDR突变体的抗体特性,以识别罕见的共同优化变体。作者团队报告了一种综合实验和计算方法,结合深度测序、机器学习和高通量实验方法来识别共同优化的治疗性抗体变体,包括相对于亲本临床期抗体具有优越亲和力和非特异性结合组合的变体。
常规分析不能很好地预测共同优化的 emibetuzumab 变体
为了识别具有高亲和力和特异性(低非特异性结合)的emibetuzumab 变体目标,通过突变重链 CDR 中的位点设计了一个大型抗体文库(约 107 个变体),接下来将文库作为单链 Fab 片段展示在酵母表面,并通过针对抗原 (HGFR) 的磁激活细胞分选(MACS,第 1-2 轮)对文库进行分类,以去除片段化或非展示抗体。然后通过荧光激活细胞分选(FACS,第 3 轮)对 MACS 分选的文库进行分选,以获得高水平的抗原结合以及与两种多特异性试剂。最后,对输入文库和 FACS 分类文库进行了深度测序,并选择了 4000 个在亲和力和特异性选择中观察到的最常观察到的抗体突变体,以进行进一步分析。
为了评估预测具有高亲和力和低水平非特异性结合的抗体突变体的能力,接下来对来自 FACS 分选文库的 125 个突变体进行了测序,并评估了它们在酵母表面上的抗原和非特异性结合的相对水平。观察到抗原结合和频率之间缺乏统计学上显着的正相关性。此外,作者还观察到对于负非特异性结合选择,非特异性结合和频率之间缺乏统计学上显着的负相关性。虽然我们确实观察到非特异性结合和富集率之间存在显着的负相关,但缺乏相应的亲和力显着相关性阻碍了使用富集率来可靠地识别高亲和力和特异性均最佳的抗体变体。
接下来,作者团队通过分析阳性类别中相对于阴性类别的文库突变的富集来评估选择的 4000 个序列数据集中包含的信息(图 2),注意到野生型残基对高亲和力和高非特异性结合选择的强烈富集。
图2 分选的 emibetuzumab 文库中的 CDR 残基富集水平与高亲和力和高非特异性结合选择相似
为了保留这些信息用于模型开发,作者团队选择将抗体 V H序列编码为 one-hot 编码向量,以捕获库中每个位点是否存在突变。假设学习这些单个特征权重的体系结构的分类算法不仅可以准确预测属性类别,还可以准确预测连续属性值。为了检验这一假设,评估了线性判别分析 (LDA) 模型预测抗体亲和力和特异性的能力(图 3)。
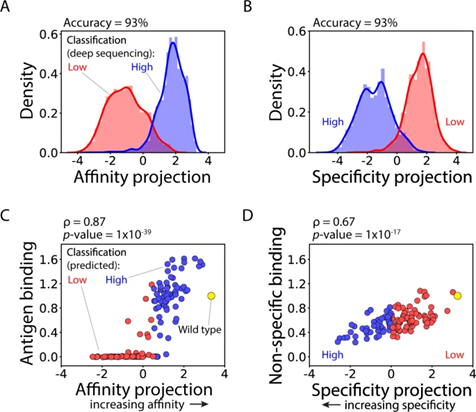
图3 (AB) (LDA) 模型使用基于序列的特征(one-hot 编码序列作为二元向量)进行训练,显示出对通过深度测序在富集文库中鉴定的 4000 种抗体的抗体亲和力和特异性进行分类的高精度。(CD) LDA 模型的连续预测,与随机选择的 125 种单链抗体 (Fabs)的相对亲和力( C )和非特异性结合( D )的实验测量密切相关。
因此,作者使用 one-hot 编码特征训练 LDA 模型以预测深度测序标签,本文称为 OneHot 模型。OneHot 模型对深度测序数据集中抗体突变体的亲和力和特异性进行了非常好的分类,两个模型(亲和力和特异性各一个)对抗体亲和力和特异性进行分类的准确率为 93%。这一发现与经验一致,并表明基于深度测序数据对抗体特性(如亲和力和特异性)进行分类是一项相对简单的任务,并且弱依赖于用于预测的模型类型。
然而,特性类别的预测对于鉴定具有最佳特性组合的抗体突变体的作用有限。但是对于LDA预测,表明不仅可用于预测类间差异(例如,低亲和力与高亲和力的分类),还可用于预测类内差异(例如,高亲和力与非常高亲和力)。因此,作者团队还评估了模型预测的能力,文库分选后通过 Sanger 测序分离(图 3C,D)。这些抗体突变体中没有一个出现在用于训练和测试的 4000 种抗体中。作者观察到模型预测和实验测量之间的强相关性,包括亲和力和非特异性指标,这些结果表明,与每个属性相关的连续指标可以以相对较高的准确度预测库中的序列。
LDA 模型的简单性引发了一个问题,即更复杂的机器学习模型是否会提高预测抗体亲和力和特异性指标的性能。因此,作者团队开发了全连接神经网络模型来预测亲和力和特异性指标。值得注意的是,神经网络模型的表现与 LDA 模型相似。两种模型的亲和力和特异性的分类预测准确度相同,即基于深度测序数据的抗体特性分类准确度弱依赖于模型复杂性。对于连续抗体特性的预测,预测抗原结合的性能相同,并且神经网络模型预测非特异性结合的性能略有提高。
作者团队接下来绘制了 LDA 模型对 4000 个抗体序列中的每一个的亲和力和特异性的预测,以在连续的尺度上直接可视化这两个属性之间的权衡(图 4A)。值得注意的是,emibetuzumab 变体在两种特性之间表现出强烈的权衡,因为亲和力的增加通常需要特异性的降低,反之亦然。
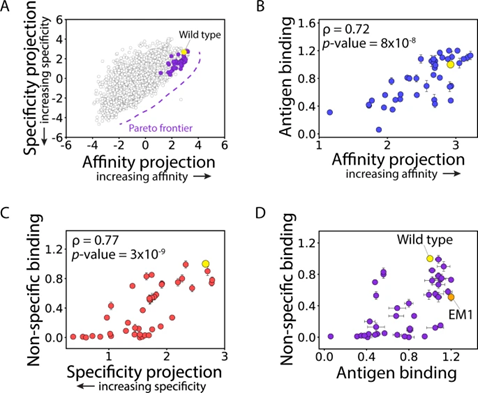
图4 分选抗体库中 emibetuzumab 突变体的帕累托最优亲和力和特异性的模型预测和实验评估。
为了评估帕累托最优抗体变体的预测,团队接下来鉴定并产生了 41 个抗体突变体,这些突变体被预测为处于或接近帕累托边界(图 4A),并通过实验评估了它们的抗原水平(图 4B)和非特异性 (图 4C) 结合,结果表明模型预测对亲和力和特异性具有很强的预测能力。
作者确定了用于进一步优化的主要候选者 (EM1),该候选者表现出抗原结合增加(1.2倍)和非特异性结合减少(0.51倍)。作者还选择了额外的克隆进行进一步的诱变,以研究优化具有多种特性的抗体突变体的潜力,试图预测新的 CDR 突变,包括以前未突变的 CDR 位点,以提高 EM1 和相关变体的亲和力和特异性。
因此,作者团队评估了基于抗体VH结构域的另外两组分子特征,以整合到预测新突变对抗体亲和力和特异性的影响的模型中。第一组特征是统一表示 (UniRep) 特征,它是从神经网络获得的深度学习特征,该神经网络在超过 2000 万个未标记的蛋白质序列上进行训练,以执行下一个氨基酸预测。第二组特征(PhysChem),称之为物理化学特征,是基于 V H结构域序列的 26 个物理化学特征。
作者团队接下来使用 UniRep 和 PhysChem 特征构建 LDA 模型,用于预测抗体亲和力和特异性指标。结果表明对抗体亲和力进行分类的准确度很高,PhysChem 模型(85%), UniRep 模型(91%),抗体特异性进行分类的准确度很高(两种模型均为 92%);同样还在两个模型上评估预测,结果表明模型预测与亲和力和特异性的实验测量密切相关。我们还使用神经网络模型重复了这一分析,简单 (LDA) 和更复杂的 (神经网络) 模型都能够预测与抗体特性密切相关的连续指标。基于两个简单 (OneHot) 特征集,这些特征集仅限于观察到的突变,而测序库和更复杂的(PhysChem 和 UniRep)特征集,可用于预测新的突变特征。受到这些结果的鼓舞,接下来直接测试我们的模型是否可以推广到新的突变空间(图 5)。
为了测试这些预测,生成了 29 种抗体变体作为可溶性 IgG,并评估了它们的亲和力和非特异性结合的相对水平(图 5),总体而言,这些发现表明,与使用传统物理化学抗体特征训练的 LDA 模型相比,使用深度学习特征训练的 LDA 模型在泛化到新的突变空间方面更胜一筹。更一般地说,这些发现证明了使用这些方法预测新 CDR 位点的抗体突变的巨大潜力,这些突变共同优化了与治疗性抗体性能相关的多种特性。
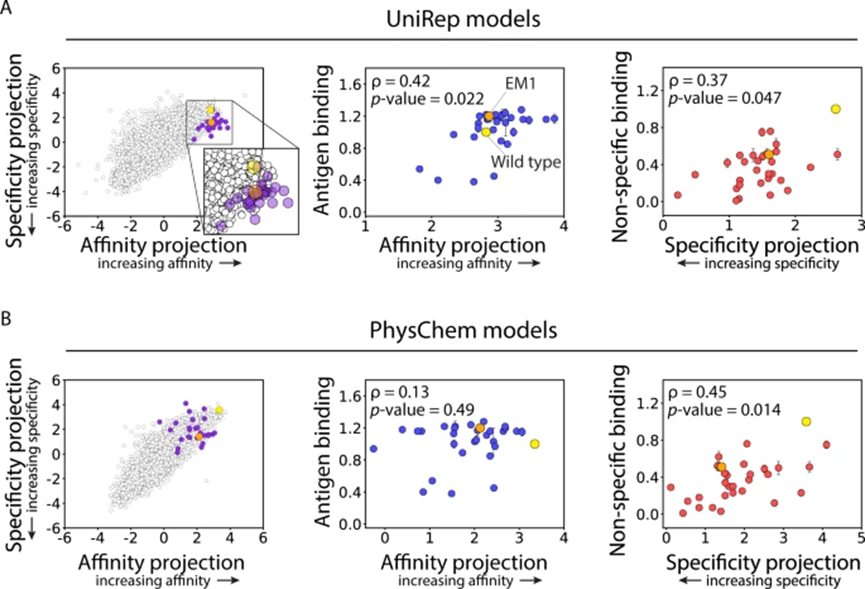
接下来绘制了本研究中产生的 70 种 IgG 的相对抗体亲和力和非特异性相互结合的实验测量值,包括原始文库中不存在的 29 种具有新突变的 IgG,以鉴定具有最多共性的变体。
EM1和 EM2(一种变体)具有比野生型更高的亲和力(图 6B)。尽管这些变体的亲和力增加,但与野生型相比,它们都显示出非特异性结合减少(图 6C),这与使用卵清蛋白获得的类似非特异性结合测量结果一致(图 6A ),此外,EM1 和 EM2 在抑制肝细胞生长因子诱导的人类癌细胞增殖方面至少与野生型抗体一样有效(图 6D)总的来说,这些结果证明了使用机器学习来共同优化治疗性抗体以提高亲和力和特异性,同时保持高生物活性和其他类似药物的生物物理特性的巨大潜力。
图6 亲和性和特异性协同优化的Emibetuzumab抗体突变体也显示出很高的生物活性和稳定性
作者团队开发了一种基于机器学习的方法,用于简化临床阶段抗体的抗体协同优化,模型表现出亲和力和非特异性结合两个特征之间具有强烈权衡,该方法还可以同时选择特定水平的多种抗体特性,从而比以前更好地控制抗体工程过程。并且本文使用深度学习训练的模型能够预测新的抗体突变,从而协同优化亲和性和特异性,还能够识别原始抗体库中不存在的有益突变,从而能够外推到新的突变空间。未来工作如过从更密切相关的蛋白质序列(例如人类抗体库)中提取深度学习特征将会带来更好的模型性能,这些模型可以越来越多地推广到新的突变空间,并减少获得共同优化的药物样抗体所需的实验量。
Makowski, E.K., Kinnunen, P.C., Huang, J. et al. "Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space." Nat Commun 13, 3788 (2022).
https://doi.org/10.1038/s41467-022-31457-3










