1. 南开黄兴禄教授AM: 基于可解释机器学习预测和设计纳米酶

大量的纳米材料被发现具有类似酶的催化活性,因此被称为纳米酶。研究表明,多种内外部因素均会影响纳米酶的催化活性。然而,目前仍缺乏必要的方法来揭示纳米酶特征和类酶活性之间的隐藏机制。
在此,南开大学黄兴禄教授等人展示了一种利用机器学习算法来理解粒子-性质关系的数据驱动方法,从而实现对纳米酶表现出的类酶活性进行分类和定量预测。为了建立纳米酶数据库,作者从300多篇论文中提取了有关纳米材料内在类酶活性的数据。在数据库建立之后,接下来需开发2种具有不同预测目标的机器学习模型:i)酶模拟类型(分类模型);ii) 类酶活性水平(定量模型)。为此,作者选择了基于深度神经网络(DNN)的全连接模型,因为它们能够处理非结构化、未标记和非线性数据。为了预测纳米酶,首先将因子作为自变量填充到输入模块中。随后,利用分类模型输出酶模拟类型(即因变量)。对于定量模型,通过标准化酶活性系数将每种类酶活性水平设置为因变量。此外,作者还引入了模型和验证数据集的正则化以克服模型的潜在过度拟合。
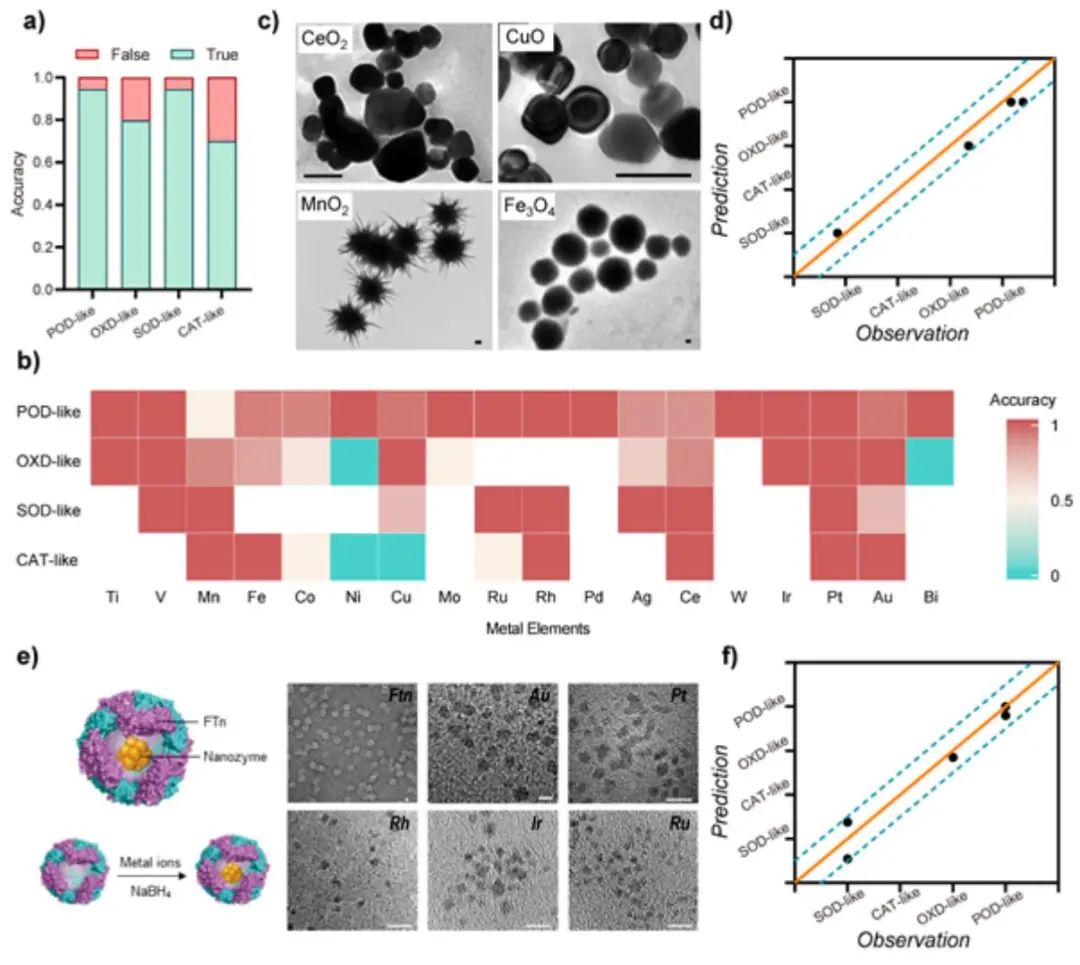
图1. 酶模拟类型的预测与实验验证
作者采用预测和观察之间的准确性来确定上述模型的性能,结果证实了该模型的高精度,显示出90.6% 的准确率。此外,Kappa一致性检验系数(高达0.803)也揭示了预测输出和观察结果之间的高度一致性。基于对模型的敏感性分析,作者揭示了过渡金属在确定纳米酶活性方面的核心作用。通过对建立的模型进行数据挖掘,可以准确预测出具有高类过氧化物酶(POD)活性的Ru基纳米材料和具有高类氧化酶(OXD)活性的Mn基纳米材料。总之,这项研究为开发具有理想催化活性的纳米酶提供了一种有前景的策略,并展示了机器学习在材料科学领域的潜力。此外,作者建议建立纳米酶评估方法的标准流程并开发更多具有类酶活性的纳米材料,以实现对具有理想功能的纳米酶设计的准确指导。
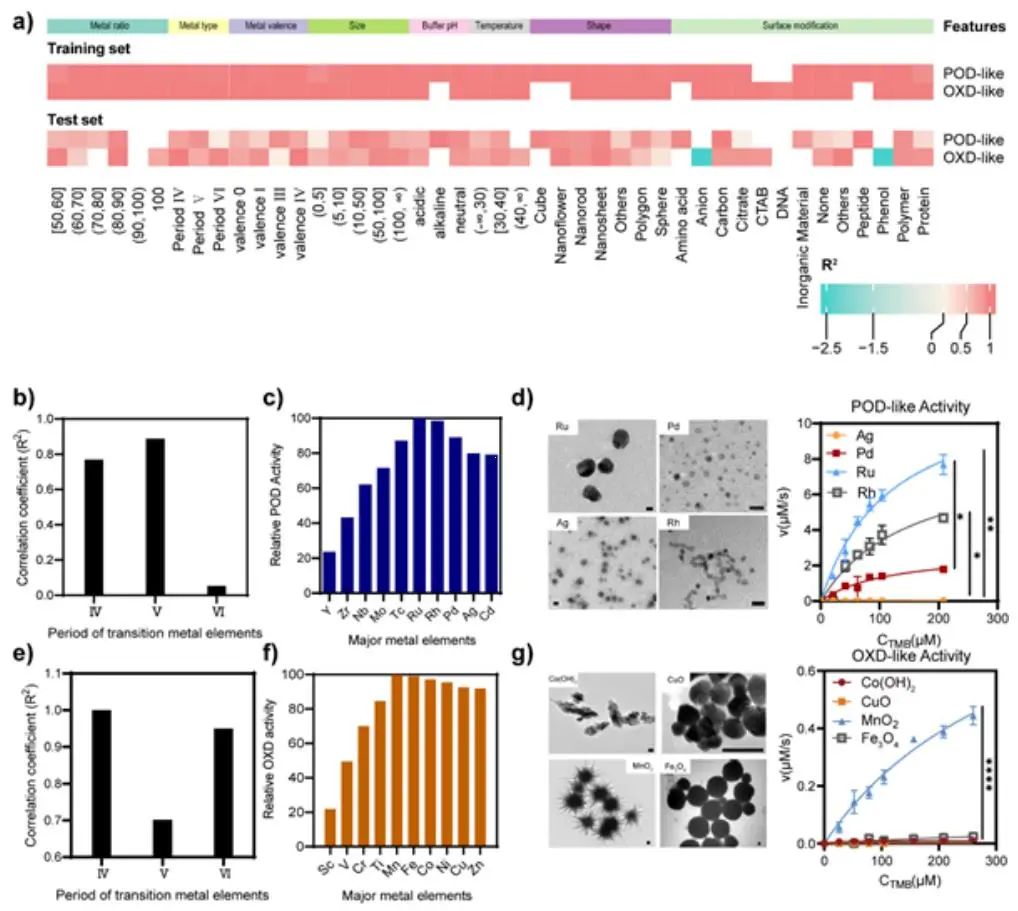
图2. 基于模型预测的金属纳米酶设计
Prediction and Design of Nanozymes Using Explainable Machine Learning, Advanced Materials 2022. DOI: 10.1002/adma.202201736
2. 李巨/熊瑞EnSM: 深度学习基于不确定的未来条件实现电池衰减预测

准确的衰减轨迹和未来寿命是新一代智能电池和电化学储能系统的关键信息,仅使用少数已知的历史数据来获得针对不确定应用条件的准确预测是非常具有挑战性的。
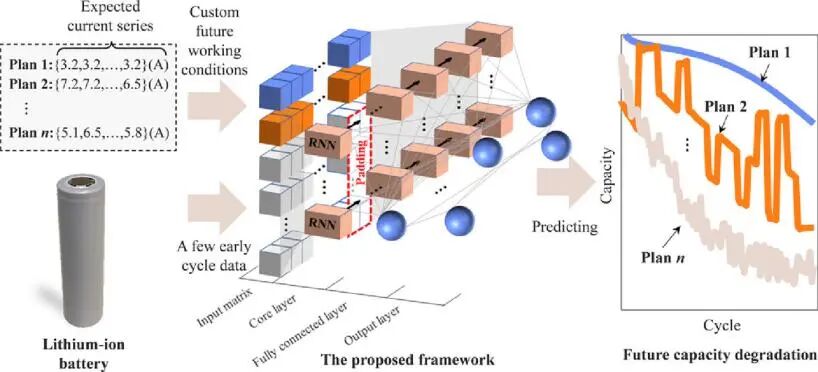
在此,北京理工大学熊瑞教授、美国麻省理工学院李巨教授等人开发了一个以循环神经网络(RNN)为核心的通用深度学习框架,从而将广泛研究的剩余使用寿命(RUL)预测扩展到固定和随机未来运行条件下的充放电容量轨迹的预测。首先,作者基于77个商用锂离子电池实验获得了涵盖固定和随机运行条件的大型电池衰减数据集(https://data.mendeley.com/datasets/kw34hhw7xg/2),所有这些电池都循环到各自的寿命终止(EOL)。这也是第一个电池衰减数据集,可适应每个电池的巨大变化的工作负载。然后,作者通过整合未来的当前计划和少量早期容量电压数据作为输入来预测固定和随机未来运行条件下的电池容量衰减轨迹。其中,该深度学习框架由四个过程组成:输入矩阵、RNN层(核心层)、全连接(FC)层和输出层。
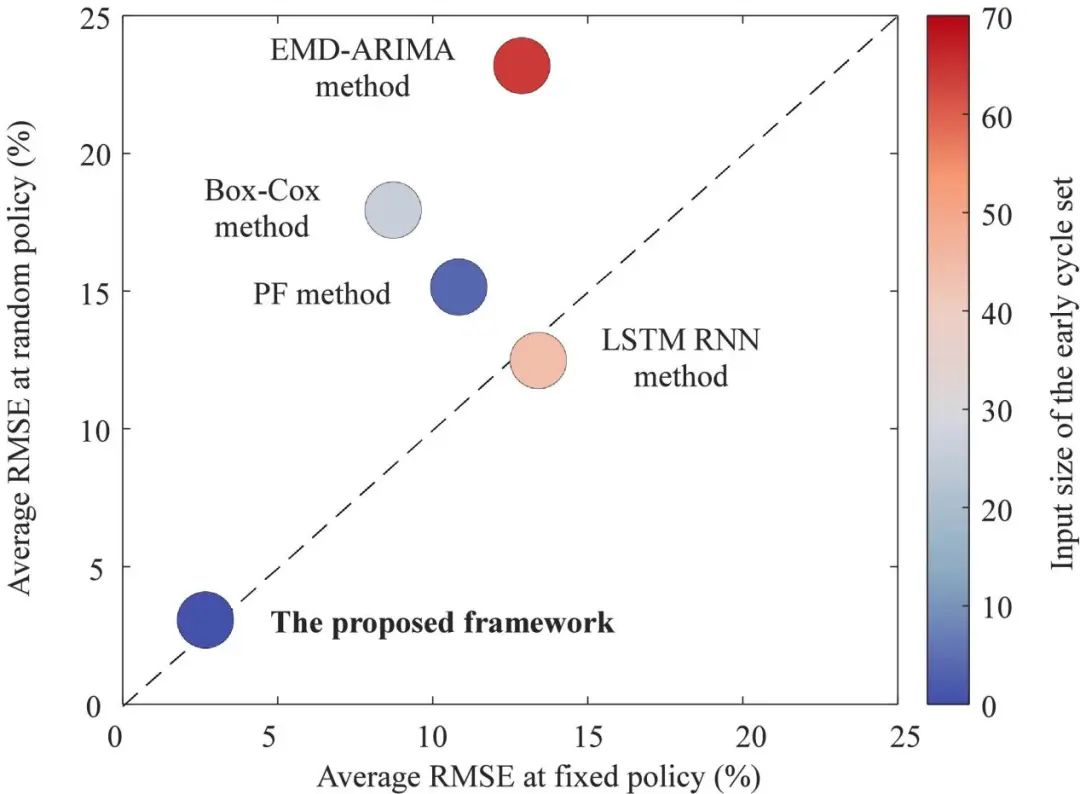
图1. 该框架与现有方法在固定和随机条件下的容量预测比较
结果表明,仅使用3.8% 的全寿命数据,NMC/石墨电池的中值预测RMSE在 2.4%以内,LFP/石墨电池的中值预测RMSE在2.3% 以内。与现有方法相比,所提出的深度学习框架预测更准确,且在固定和随机未来条件下都具有令人满意的性能。尽管如此,该研究仍可在未来得到改进:首先,所提出的框架不限制输入特征维度,因此可探索框架以应用于更通用的随机策略,如温度、放电深度或其他导致衰减的因素;其次,所提出的框架应通过一些实验数据进行训练,因此可进一步探索这种通用框架在迁移学习中的潜力;最后,可探索提出的框架以应用于大数据,引入涵盖历史真实世界数据分布的大数据可能会导致更正确和有效的学习。总之,该研究提出的框架有望帮助电池快速开发和定制电池管理软件以延长电池寿命,并有助于对电池组中的许多电池单元进行智能控制。
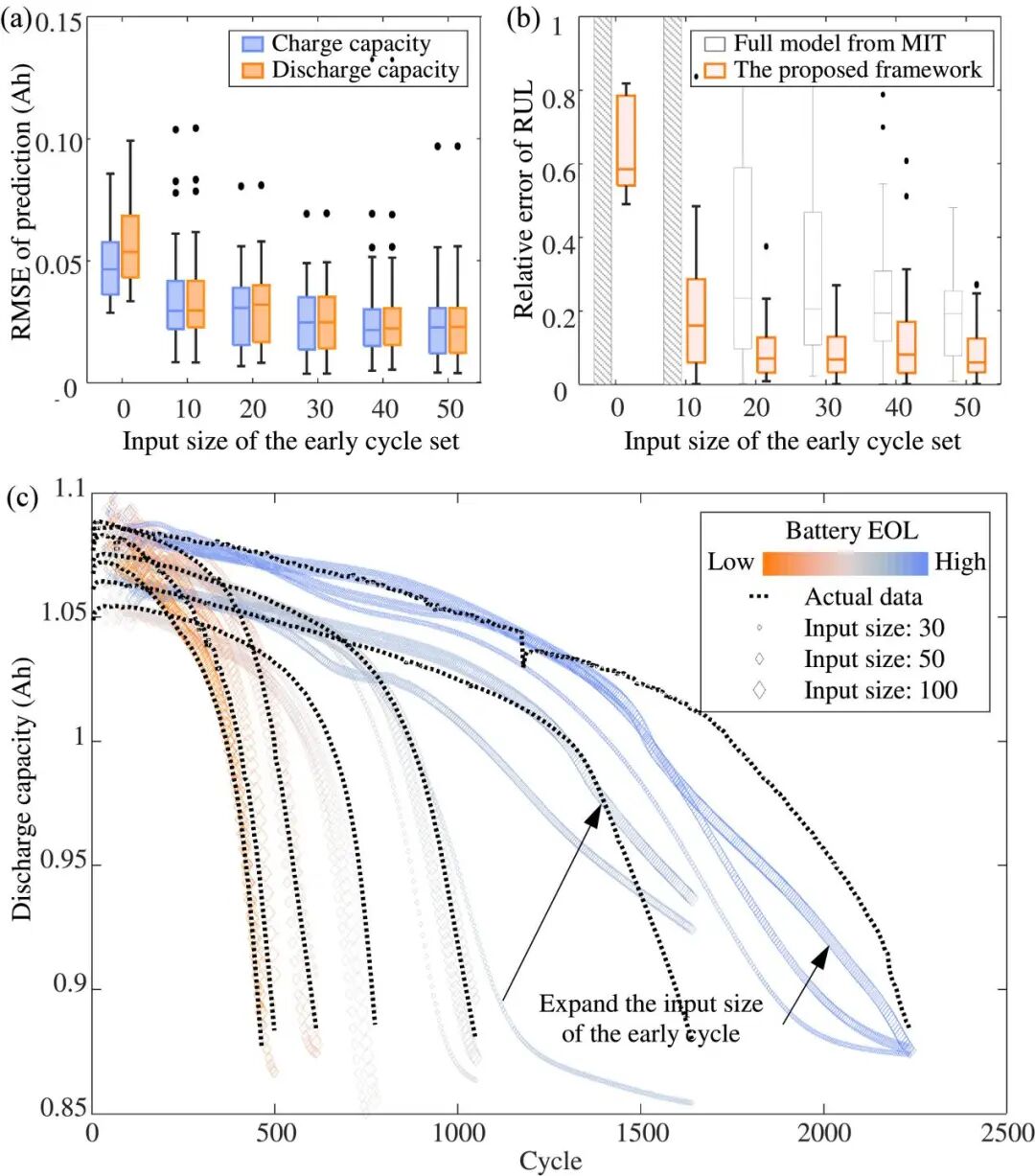
图2. 该框架基于LFP/石墨电池的容量衰减预测性能
Battery degradation prediction against uncertain future conditions with recurrent neural network enabled deep learning, Energy Storage Materials 2022. DOI: 10.1016/j.ensm.2022.05.007
3. 复旦刘智攀/商城ACS Catal.: 机器学习模拟研究固-气/液条件下CO氧化

自由能势垒及其温度依赖性和化学反应的活化熵是反应动力学的关键量,但其准确预测非常具有挑战性,特别是对于复杂环境中的反应。
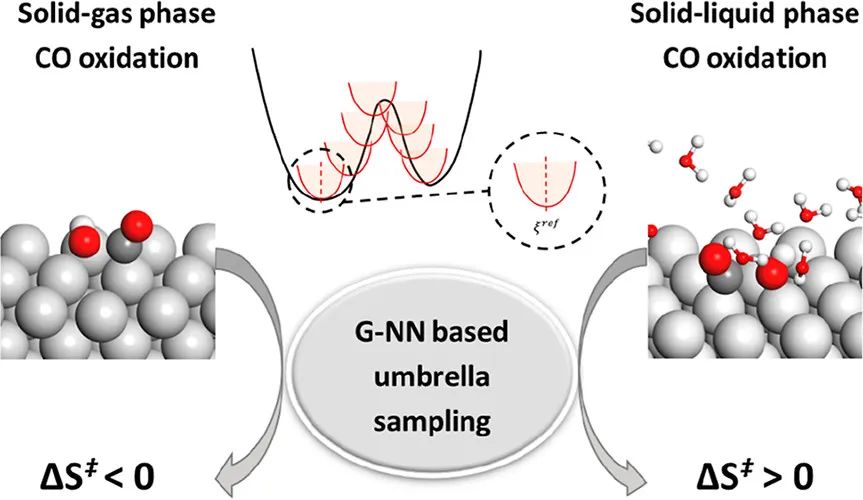
在此,为了解决长期以来在不同反应条件下CO氧化的难题,复旦大学刘智攀教授、商城研究员等人通过基于机器学习的原子模拟系统地研究了在不存在和存在水溶液的情况下Pt(111)上CO的氧化。其中,作者通过将全局神经网络势(G-NN)与伞形采样分子动力学(MD)相结合来计算CO与原子O和OH在固-气和固-液环境中反应的自由能分布,即CO + O(g)、CO + OH(g)、CO + O(aq)和 CO + OH(aq)(g和aq分别代表气体和水溶液)。G-NN势遵循原子中心高维神经网络结构,并通过自学习随机表面行走(SSW)全局优化筛选出来的全局势能表面(PES)来生成数据集,最终数据集包含由DFT计算的79724个结构。此外,G-NN采用幂型结构描述符(PTSD)作为神经网络的输入层,每个单元由170个二体、200个三体和40个四体PTSD组成。
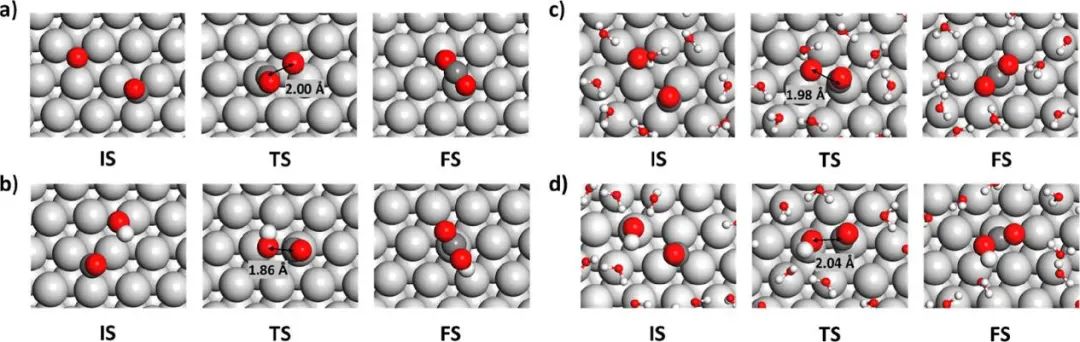
图1. MD模拟的反应快照
通过分析MD轨迹以观察构型、平移和旋转自由度,作者揭示了活化熵的物理起源。本文研究结果总结如下:(1)在低覆盖极限的固气条件下,CO+O的自由能势垒为0.96 eV,CO + OH反应在300 K时的自由能势垒为0.46 eV。两个反应的活化熵均为负值,分别为-12.28和-20.88 J mol-1 K-1;(2)在低覆盖极限的水溶液中,CO + O的自由能势垒为0.91 eV,300 K时CO + OH反应的自由能势垒为0.76 eV。两个反应的活化熵均为正值,分别为20.57和31.80 J mol-1 K-1。在电化学条件下,CO + OH反应仍主导Pt电极上的CO氧化;(3)活化熵通常远大于从谐波近似计算的振动熵,表明构型熵在表面反应的活化熵中占主导地位;(4)CO氧化活性对CO覆盖率非常敏感。总之,这项研究为不同反应条件下的 CO 氧化提供了新的物理见解,对理解和预测催化重组反应的自由能势垒具有重要意义。
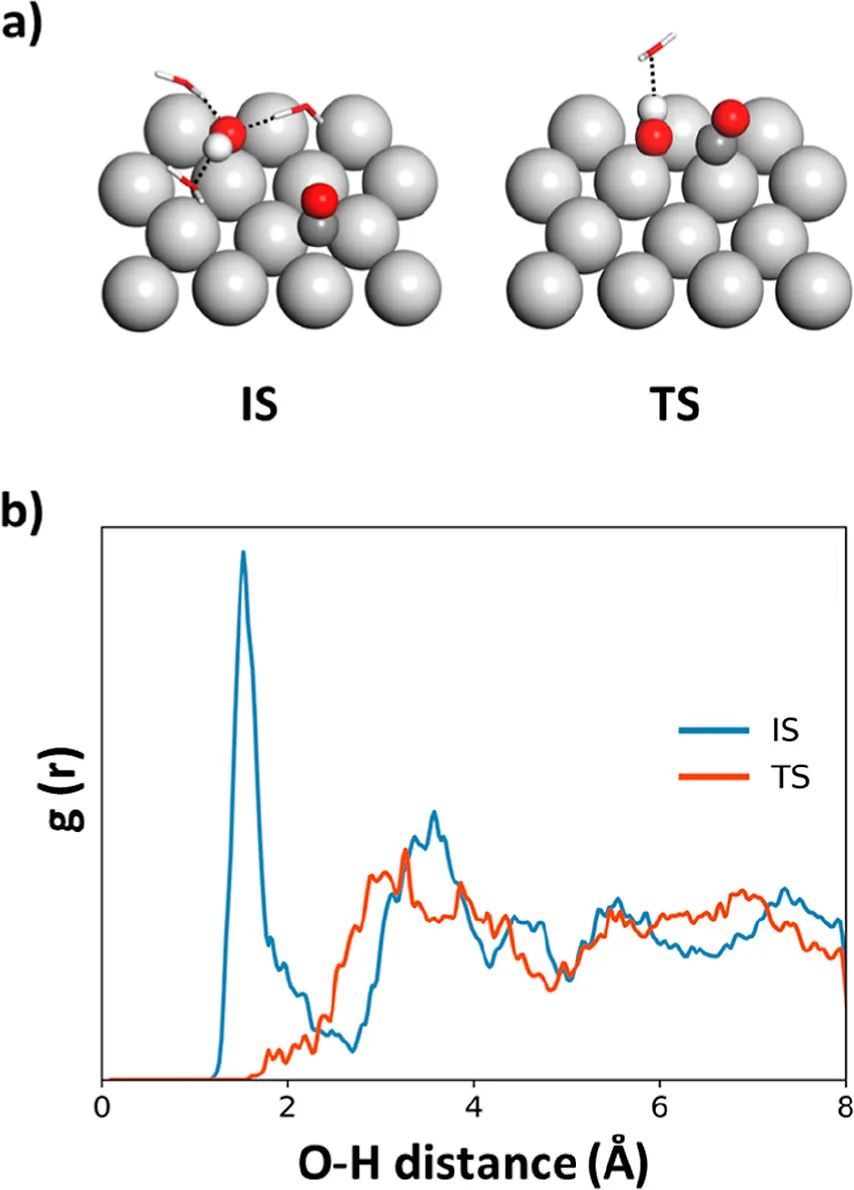
图2. 水溶液中额外的氢键稳定吸附的OH
Resolving Activation Entropy of CO Oxidation under the Solid–Gas and Solid–Liquid Conditions from Machine Learning Simulation, ACS Catalysis 2022. DOI: 10.1021/acscatal.2c01561
4. Chem. Sci.: 通过无机材料的对比表示学习克服缺乏训练数据集的挑战!

数据表示形成了一个特征空间,其中形成了数据分布,这是决定机器学习(ML) 预测准确性的关键因素之一。特别是,数据表示对于处理小型和有偏差的训练数据集至关重要,这是ML在化学应用中的主要挑战。
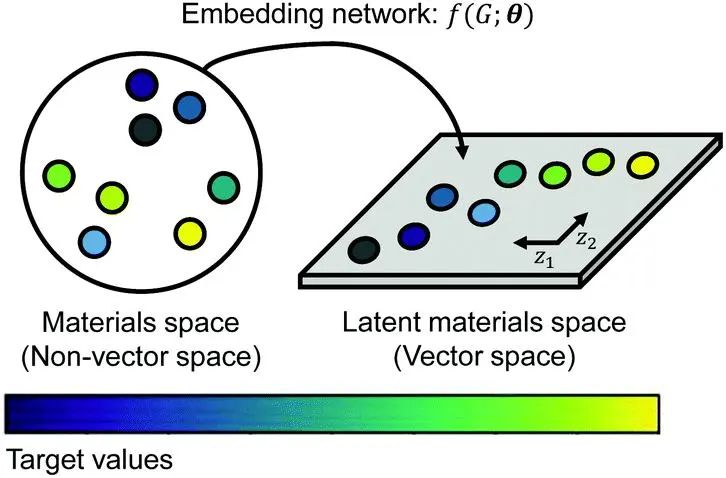
在此,韩国化学技术研究院Gyoung S. Na、光州科技学院Hyun Woo Kim提出了一种与数据无关的表示方法即显式材料表示学习(EMRL),可自动且普遍地生成目标材料特性的晶体结构的最佳数据表示。作者展示了EMRL嵌入网络的嵌入空间(潜在材料空间)的概念,即通过嵌入网络将非向量空间(原始材料空间)中的材料投影到向量空间(嵌入空间)中,从而更容易处理数据。在生成的嵌入空间中,材料根据其目标属性的值进行粗略排列,这使得回归问题更容易。作者评估了基于EMRL的ML算法的预测性能,以验证EMRL的有效性。具体而言,作者生成了一个梯度提升树模型EMRL-GB并以EMRL生成的材料表示作为预测材料属性的输入,共进行了4次实验来预测小型训练数据集、有偏差的训练数据集和迁移学习环境的材料属性。
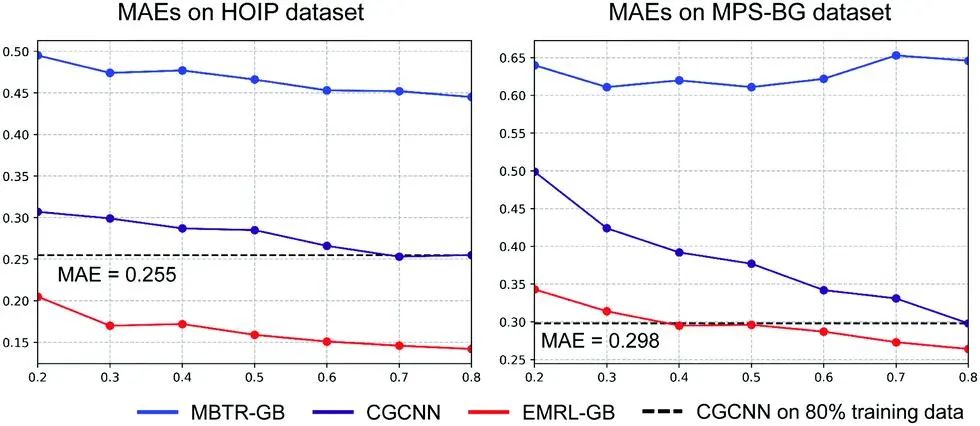
图1. 不同模型基于两种数据集上不同比例训练数据的预测误差
结果显示,EMRL-GB仅使用HOIP数据集上20%的训练数据,便实现了与晶体图卷积神经网络(CGCNN)在80%数据上的可比预测误差。对于MPS-BG数据集,基于30%训练数据的EMRL-GB也表现出与CGCNN在80%数据上训练误差相当的预测性能。此外,在预测未知材料组中材料物理性质的外推问题中,基于EMRL的ML算法预测精度提高了28.89~30.87%,EMRL的源代码可在 https://github.com/ngs00/emrl/tree/master/EMRL公开获得。作者将EMRL在材料科学的ML应用中的优势总结如下:1)针对给定材料目标特性自动且普遍地生成晶体结构的最佳表示;2)可提高ML算法在外推和知识转移的回归问题中的预测能力;3)对于发现新材料很有用,因为它提供了通用且稳健的材料表示,这对于探索未知材料空间至关重要。
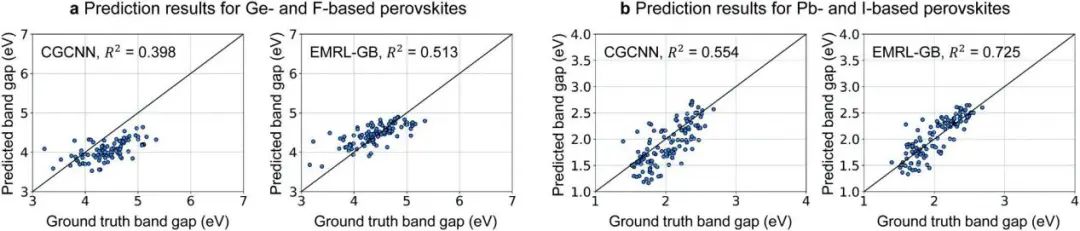
图2. 基于EMRL的ML算法在有偏差数据集上的预测结果
Contrastive representation learning of inorganic materials to overcome lack of training datasets, Chemical Communications 2022. DOI: 10.1039/D2CC01764D
5. 华东理工孙辉教授CEJ: 机器学习预测用于硫化羰化学吸收的溶剂识别

由于通过实验或计算方法对反应动力学的研究具有挑战性,因此探索用于硫化羰 (COS)化学吸收的溶剂在很大程度上受到阻碍。目前,机器学习(ML)已被证明可极大地加速学术和工业领域的信息识别和功能实现。
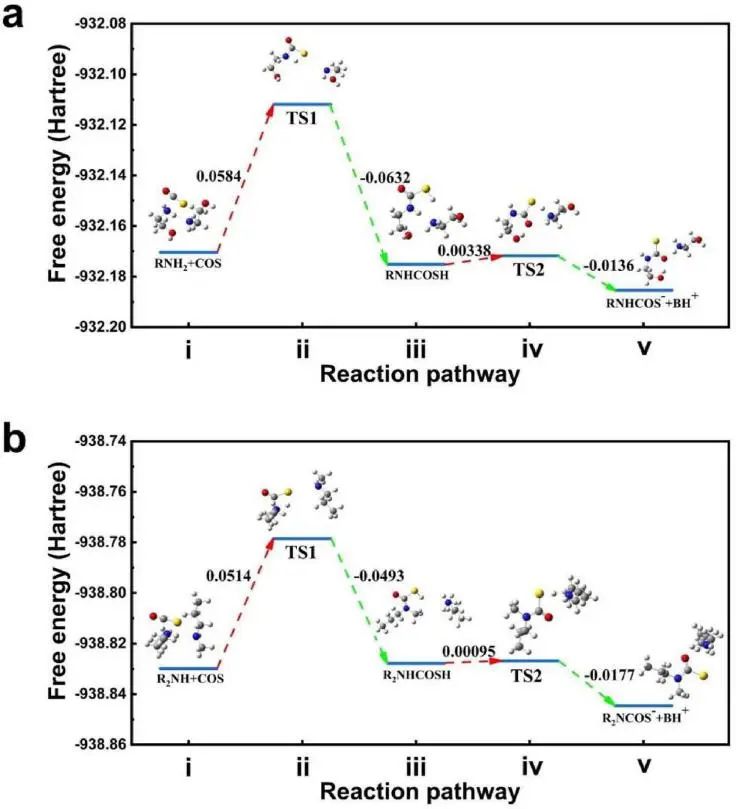
在此,华东理工大学孙辉教授等人报道了一种机器学习(ML)引导的反应动力学预测,用于智能识别有前景的COS化学吸收化合物。首先,作者识别了COS 的溶剂参与反应的速率决定步骤,并通过量子化学计算获得了决速步骤的反应速率常数以建立初始训练数据集。此外,作者定义了代表胺基周围空间位阻的两个分子描述符alphaV和betaV来构建ML算法,并使用SHapley Additive exPlanations(SHAP)方法来解释模型。鉴于预测精度高、可解释性好、避免过度拟合的泛化能力强,作者构建了一种集成学习方法,即随机森林(RF)来预测COS与潜在溶剂化合物反应的速率常数。数据集被随机分成训练数据集(80%)和测试数据集(20%),并以计算的三个性能指标,包括测试集的相关系数(Q2)、均方根误差(RMSE)和平均绝对误差(MAE)来评估ML模型的性能。
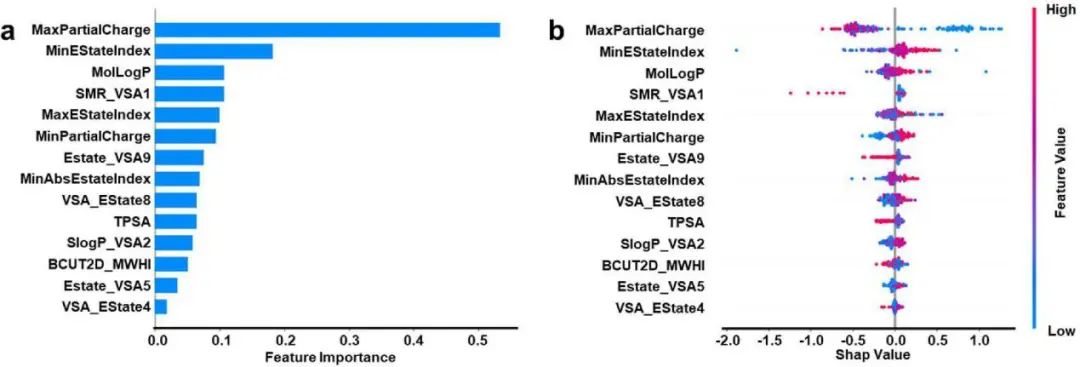
图1. 描述符的特征重要性和SHAP分析
在引入四个特定描述符后,包括自定义的alphaV、betaV描述符及MaxNCharge和MaxEState_N的另外两个关于胺基电子特性的描述符,ML模型预测反应速率常数的MAE大大降低,其值为0.80。构效关系表明,反应速率很大程度上取决于溶剂化合物的电荷分布和空间位阻。作者确定了一个合理的胺基电子密度范围,在这个范围内COS的反应可以达到促进的反应速率。相反,反应可被α-C上的空间位阻严重抑制。此外,作者使用该ML算法预测的反应速率常数与5种商业胺化合物的实验结果进行了比较,所有实验数据与计算结果吻合良好,表明该ML 模型对于智能预测COS与溶剂候选物的反应动力学是有效的。总之,本研究为ML引导探索和识别通过化学吸收过程进行化合物捕获和分离的溶剂候选物提供了通用策略。
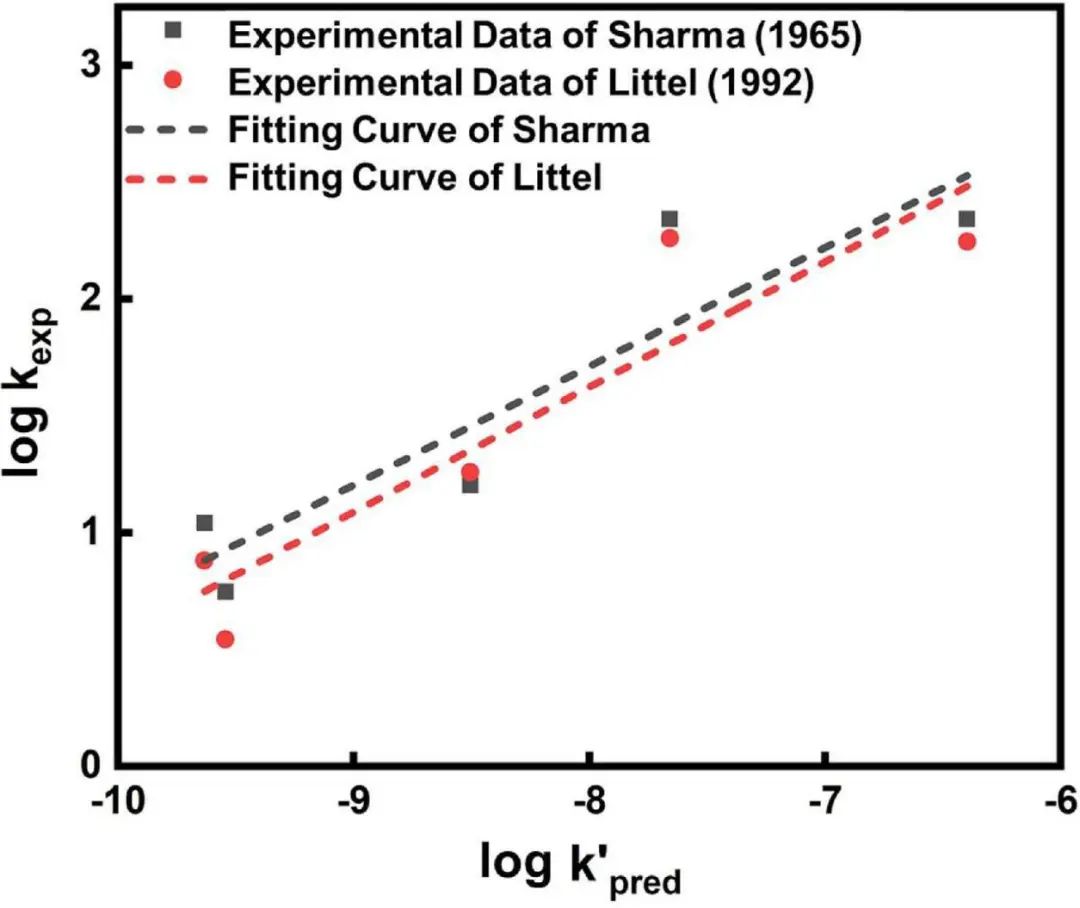
图2. ML算法预测的反应速率常数与实验结果的比较
Machine-learning-guided reaction kinetics prediction towards solvent identification for chemical absorption of carbonyl sulfide, Chemical Engineering Journal 2022. DOI: 10.1016/j.cej.2022.136662
6. 河南农大/登嘉楼大学CEJ: 机器学习用于生物质水热液化的预测和优化

水热液化过程最近在生物精炼厂的设计和实施中引起了更多的关注,因为它能够处理各种湿性生物质原料。然而,测量水热液化(副)产物的定量和定性特征具有挑战性,因为需要进行耗时且成本密集的实验。机器学习(ML)技术能够从过去的数据集和机制中学习,因此可用于解决上述问题。
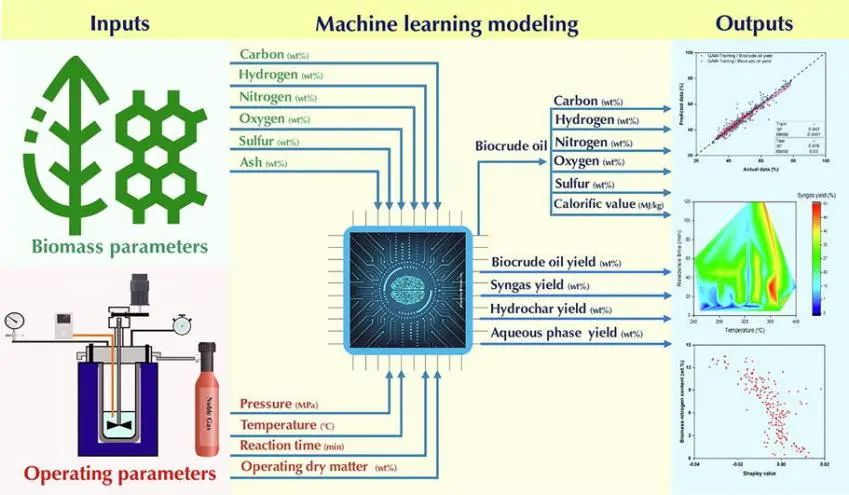
在此,河南农业大学Mortaza Aghbashlo副教授、马来西亚登嘉楼大学Su Shiung Lam教授及Meisam Tabatabaei等人应用机器学习(ML)来基于生物质组成和反应条件定量和定性地表征其水热液化(副)产物。首先,作者对已发表的有关该主题的文章进行了检索和筛选,并从符合条件的研究文章中提取所需的数据。然后手动删除不合理的数据,并对收集的数据进行统计分析和机械讨论。作者采用了4种ML模型,包括神经网络回归(NNR)、广义加性模型(GAM)、支持向量回归(SVR)和高斯过程回归(GPR)用于对上述过程进行建模。使用进化算法,即粒子群优化(PSO)对ML模型的超参数进行调整,然后将性能最佳的ML模型开发的目标函数输入多目标进化算法,即多目标粒子群优化(MOPSO),以找到最佳的生物质组成和反应条件。
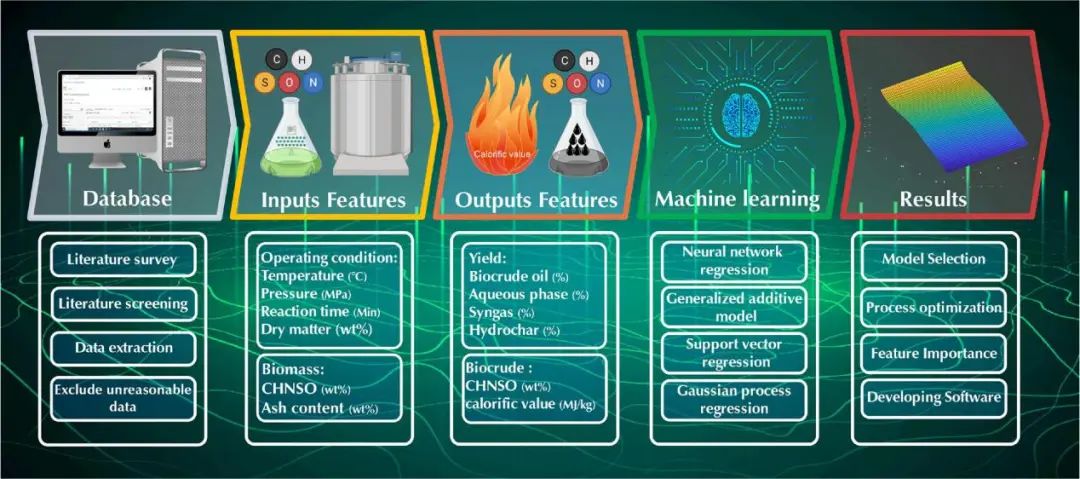
图1. 本研究中使用的研究方法
结果表明,在所考虑的4种ML模型中,GPR模型可以提供最高的准确度,R2高于0.926,平均绝对误差低于0.031。使用所选模型开发的目标函数可最大限度地提高生物原油的数量和质量,并最大限度地减少副产品的数量。当生物质中碳、氢、氮、氧、硫、灰分含量分别为40.9 ~ 48.3%、9.72 ~ 9.80%、11.9 ~ 13.6%、15.2 ~ 15.6%、0.0 ~ 0.94%、0.0 ~ 2.92% 时,生物原油产率最佳为48.7 ~ 53.5%。其中,最佳操作条件为:干物质含量为31.4 ~ 33.0%,温度为 394 ~ 400℃,反应时间为5 ~ 9 min,压力为30.0 ~ 35.6 MPa。最后,作者将性能最佳的GPR模型嵌入到用户友好的软件包中,以预测生物质水热液化(副)产品的质量和数量。这项研究开发的软件包可有效地消除对耗时且成本高昂的实验试验的需求,并促进包括生物质水热液化工厂的生物精炼系统的设计、分析、优化和改造。
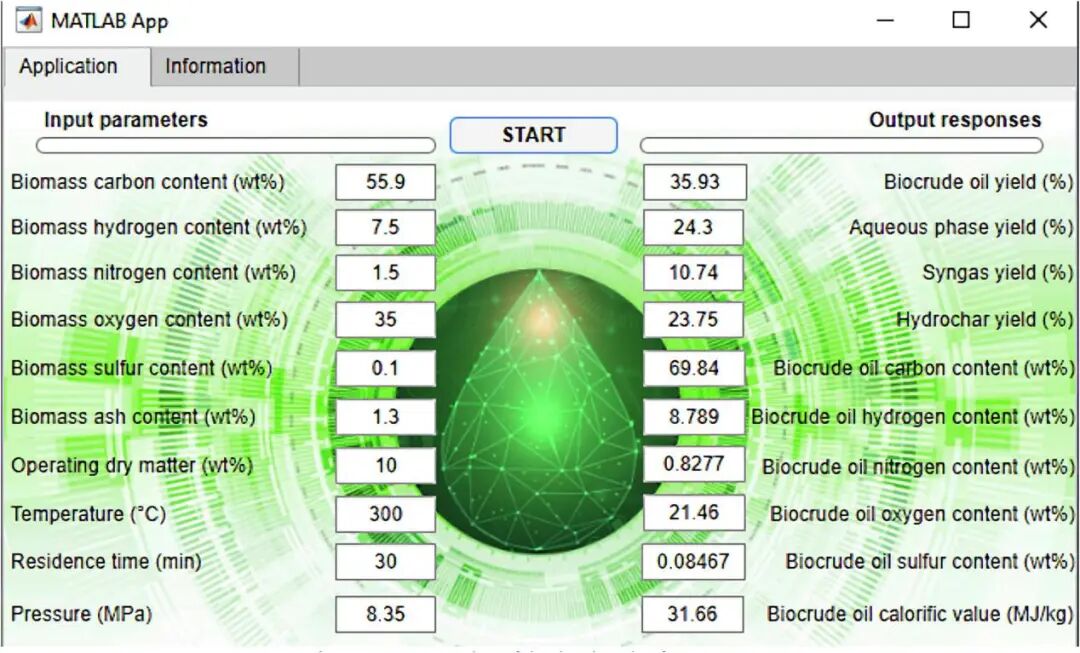
图2. 开发软件的界面截图
Machine learning predicts and optimizes hydrothermal liquefaction of biomass, Chemical Engineering Journal 2022. DOI: 10.1016/j.cej.2022.136579

球差、冷冻电镜、原位TEM、HR-TEM、EDS-Mapping、SAED、HADDF-STEM、EELS、ABF-STEM应有尽有!Science、Nature级别水平!发顶刊,拍TEM,找我们就对了!添加下方微信好友,咨询宋老师:13612927057(电话同微信)










