位点特异性重组(SSR)是生物基因重组的一种机制,也是合成生物学中的重要工具。该过程涉及使用识别和修改活细胞中特定 DNA 序列的酶,因此在使用细胞疗法治疗疾病方面有重要应用。
例如,免疫疗法需要从患者体内提取免疫细胞并对其进行基因改造以对抗癌症等疾病。在这些应用中,重要的是精确控制基因表达的时间,以最大限度地提高治疗效果,同时最大限度地减少体内的不良反应。
然而,SSR 反应速率仍受限于无法进行简单且可预测地跨数量级的调整,简单的速率操作原则上可以通过修饰重组酶的 DNA 底物的核苷酸序列来实现,但这种方法的设计原则尚未阐明。
为了能够通过 DNA 序列对 SSR 反应动力学进行可预测的调整,近日,明尼苏达大学双城分校的研究人员开发了一种方法,将高通量实验与机器学习模型相结合,使特定位点的重组过程更有效和可预测。该模型允许研究人员对基因编辑的速度进行编程。这意味着它们可以控制工程化细胞对环境的反应速度,从而控制它产生治疗性蛋白的快慢。相关研究以“Model-guided engineering of DNA sequences with predictable site-specific recombination rates”为题发表在 Nature Communications 上。

(来源:Nature Communications )
该论文的通讯作者、明尼苏达大学双子城生物医学工程系副教授 Casim Sarkar 表示,据我们所知,这是第一个使用模型来预测修改 DNA 序列如何控制位点特异性重组率的例子,通过植入工程思想,我们可以调节 DNA 编辑发生的速度,并使用这种控制形式来定制工程化细胞反应。除此之外,我们的研究还确定了比自然界中发现的更有效重组的新 DNA 序列,这可以加快细胞的反应时间。

从改变酶的关键氨基酸残基到专注设计 DNA 附着位点序列
根据已有信息,SSR 技术依赖于重组酶,重组酶可精确识别两个 DNA 位点、形成中间复合物、切割、交换和重组 DNA,从而导致基因插入、缺失或倒位。根据催化结构域中的活性残基,重组酶超家族可分为两类:酪氨酸重组酶和丝氨酸重组酶,每组可根据酪氨酸重组酶的方向性(双向/单向)或丝氨酸重组酶的大小(大/小)进一步细分。
其中,大丝氨酸重组酶(LSR)基于以下特性被认为是合成生物学中最强大的工具之一:
不可逆性:LSR 具有不同的识别位点,通常称为 attB(附着位点细菌)和 attP(附着位点噬菌体),并产生杂合产物位点 attL 和 attR。LSR 不能靶向混合 attL 和 attR 位点来再生 attP 和 attB,从而产生异常稳定的 DNA 重组产物;
简单性:与需要长 attP 位点(>200 bp)、超螺旋 DNA 结构和其他稳定 DNA 弯曲的因素的 λ 整合酶等酪氨酸重组酶相比,丝氨酸重组酶具有短 DNA 位点(<50 bp),并且不需要DNA 拓扑结构或辅助因子;
效率:Bxb1、TP901 和 PhiC31 等 LSR 已被证明在原核和真核细胞、基因治疗、 记忆电路设计、和基因组编辑中均有效。
因此,研究人员打算设计重组酶 Bxb1 的 DNA 附着序列 attP,以可预测地调节由该酶介导的反转反应。该论文阐述,此前,调节生化反应速率的工程方法集中在改变酶中的关键氨基酸残基。然而,合理的蛋白质设计受到缺乏高分辨率重组酶-DNA 复杂晶体结构的限制。尽管一直在努力了解重组酶的氨基酸残基和核苷酸之间的相互作用,但静态重组酶-DNA 复杂晶体结构无法提供足够的信息来了解 SSR 率的 DNA 序列决定因素。
除了直接的蛋白质-DNA 接触外,电荷/形状互补性和水介导的相互作用也有助于不同 DNA 序列的 SSR 率。此外,重组酶的突变会改变蛋白质的稳定性或溶解度,从而混淆通过酶工程合理调整反应速率的努力。因此,研究人员转向专注于开发一种合理设计 DNA 附着位点序列以调节 SSR 反应速率的方法。
“可为患者提供个性化治疗”
研究人员首先对 DNA 文库进行了设计,具体做法如下:首先确定了 Bxb1 的 attP 和 attB DNA 附着位点中的核苷酸位置,值得注意的是,Bxb1 的 attP 和 attB 位点具有不相同的序列,两者都有两个准半位点 attP-L/attP-R 和 attB-L/attB-R。最终因为种种原因,选择在 attP-L 半位点内引入突变;接着,研究人员构建了一个 DNA 文库,其中包含 attP-L 中从 9 到 18 的 10 个连续位置的随机碱基,包括对称位点和不对称位点。
同时,为了给调整反应速率提供更大的灵活性,研究人员还设计了一个新的饱和诱变 DNA 文库(图 1C),通过改变不对称位置(3G、5G、8G、11T、12G、14C、15C、16A、17G)来识别具有更广泛反应速率的变体,同时保留一个假定的保守碱基 19C 作为选择中的对照。
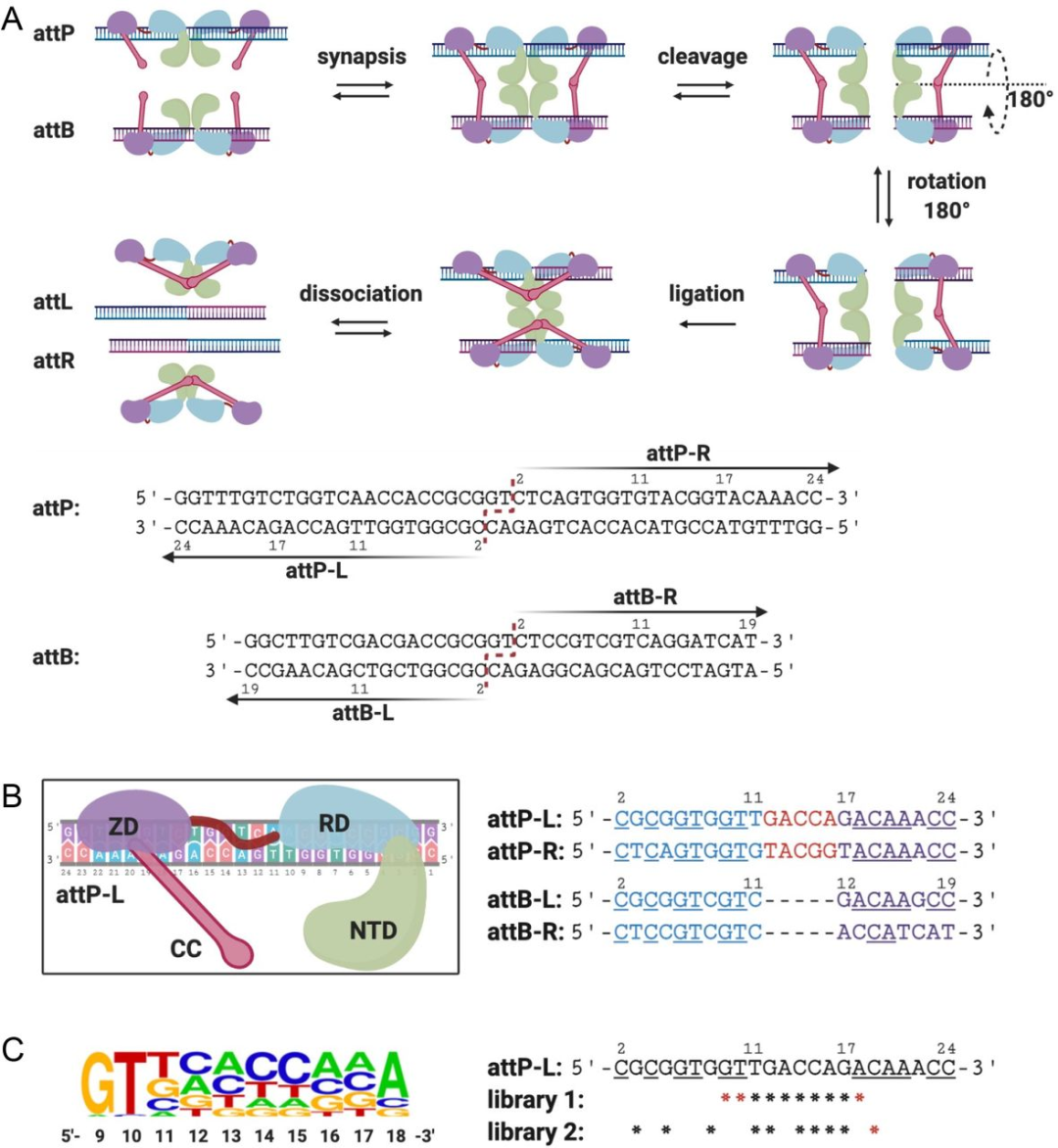
▲图1丨Bxb1 attP-L DNA 文库设计(来源:研究论文)
接着,研究人员开发了一种基于 qPCR 的方法,可通过量化重组 DNA 产物浓度来准确测量初始 SSR 反应速率。简而言之,研究人员构建了一个线性 DNA 片段,其两侧是位于相反方向的 attP 和 attB 位点。Bxb1 与 attP/attB 位点结合,然后反转侧翼 DNA 片段。研究人员设计的引物与 attP 序列上切割位点的相邻序列互补,并且反转反应改变了引物之一的方向及其退火的 DNA 链,因此 PCR 导致反转的指数扩增模板,重要的是,它包括 attP-L 序列。对于未翻转的 DNA 底物,两条引物在同一条链上以相同方向延伸,因此无法通过 PCR 进行扩增。研究人员使用 qPCR 来测量野生型(WT)attP 位点在不同 Bxb1 浓度下翻转的百分比,结果与 DNA 凝胶电泳的结果一致。
为了测试这种定量方法的灵敏度,研究人员还绘制了 Cq 值(量化周期)和翻转 DNA 模板浓度的标准曲线,结果发现,该 qPCR 方法准确地测量了翻转的 DNA 百分比,与以前基于电泳迁移率或核酸外切酶消化差异的方法相比,该方法简便易行,可以准确测量初始反应速率。同时,通过这种灵敏且准确的基于 qPCR 的速率测定,研究人员还使用 DNA 文库作为底物选择了具有高重组效率的 DNA 变体。
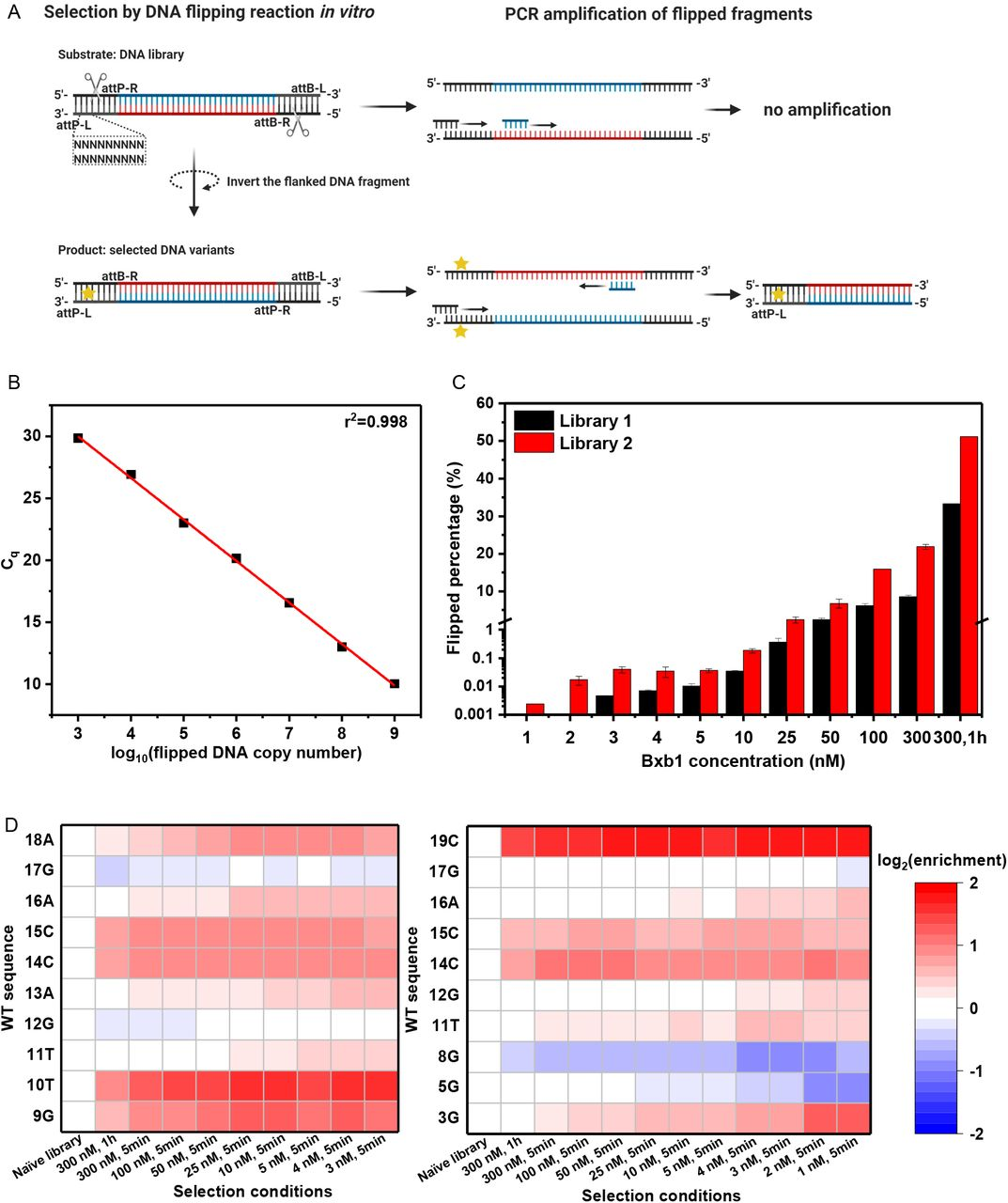
▲图丨通过 qPCR 对 DNA 倒置和 attP-L 文库选择进行量化(来源:研究论文)
总的来说,Sarkar 和他的团队首先开发了一种实验方法来计算特定位点的重组率,然后使用该信息来训练机器学习算法。最终,这使研究人员可以简单地输入 DNA 序列,该模型可以预测该 DNA 序列重组的速率。使用这种方法,通过在 attP-L 半位点序列中进行合理的核苷酸替换,可以将反应速率编程为四个数量级。(先前对位点特异性重组酶的表征研究集中在 DNA 识别的特异性上,尚未报道反应速率对 DNA 序列的依赖性的定量分析。)
其次,通过组合多个单独对重组效率进行适度改进的取代,研究人员还创建了一个新的 attP-L 序列,它使初始反应速率比 WT(野生型)提高 10 倍。除此之外,该研究团队还发现可以使用建模来预测和控制细胞内多种蛋白质的同时产生,这可用于对干细胞进行编程,以产生用于再生医学应用的新组织或器官,或赋予治疗细胞以预定比例生产多种药物的能力。
“不同的患者可能需要不同的剂量或更快或更慢的细胞反应——不是每个人都是一样的,”Sarkar 解释说。“通过在细胞内建立利用具有不同和确定重组率的多个 DNA 序列的遗传回路,我们现在可以实现以前难以做到的事情,例如治疗细胞中蛋白质生产的程序比率。我们的合理方法能够为患者提供个性化治疗。
”
1.https://www.scienceboard.net/index.aspx?sec=ser&sub=def&pag=dis&ItemID=4483
2.https://www.nature.com/articles/s41467-022-31538-3
3.https://phys.org/news/2022-07-tool-personalized-cell-therapies.html
欢迎合成生物学领域科研从业者扫码加群,加好友请备注“单位+领域+职位”(不加备注不予通过)↓↓↓








