蛋白质结构域是位于二级结构和三级结构之间的一个层次,每个结构域一般由100~250 个氨基酸残基组成,单个结构域具有独特的空间构象。一条较长的蛋白质全长单链通常会包括若干个结构域,某些区域相邻的氨基酸残基首先形成有规则的二级结构,然后由若干二级结构折叠成近似于球状的结构域,最后通过两个或多个结构域组装形成多结构域蛋白(简称多域蛋白)的三级结构。结构生物学领域普遍认为结构域是蛋白质三级结构的独立折叠单元和独立功能单元。从生物学意义上来讲,多域蛋白质中结构域间缔结对于促进以协作方式实现多个相关的功能至关重要,域间铰链区(Linker)结构柔韧性使结构域之间容易发生相对运动,这将有助于结合底物或施加应力,有助于别构中心结合调节物和发生别构效应,从而促进于酶对反应的催化,致使这些部位往往是活性中心所在的部位,或是变构物结合的部位。因此,阐明多域蛋白的结构是理解其全部功能的关键步骤!
自然界生物中,大约有超过80%的真核蛋白和67%的原核蛋白均含有多个结构域。PDB库最新统计结果表明,现有实验测定结构蛋白质中只有大约35%的单链为多域蛋白,且绝大多数的多域蛋白单链仅包含2个或3个结构域,极少数超过5个结构域。考虑到PDB库中存储的蛋白结构源于实验测定,似乎可以得到一个明显的结论:由于X衍射、NMR及冷冻电镜等结构生物学实验测定手段的技术瓶颈,多域蛋白结构实验测定速度远低于单域蛋白。幸运的是,随着人工智能、生物大数据及计算技术的深入发展,2020年谷歌DeepMind团队推出的AlphaFold2,使得蛋白质单结构域预测问题取得了突破性的进展。AlphaFold2几乎可以正确预测所有单域蛋白质结构,模型精度几乎可以与生物湿实验测定结构相媲美。但是也发现,对多域蛋白质全链建模的平均精度似乎低于建模所组成域结构的平均精度,这也正好说明了多域蛋白质结构建模技术仍然是一个目前尚未解决的挑战性研究课题。如何利用局部结构相似性与全局结构相似性的内在关系,同时在降低训练、推断的硬件需求前提下,进一步提高多域蛋白质建模精度是一个值得关注的重要问题。
浙江工业大学张贵军教授课题组于2022年8月13日在生物信息学领域期刊《Bioinformatics》上发表了题为《Structural analogue-based protein structure domain assembly
assisted by deep learning》的研究论文(图1),提出了深度学习辅助的结构类似物多域蛋白质域组装方法(简称SADA)。在356个测试蛋白上,SADA与主流的结构域组装方法进行了比较,SADA模型的TM-score分别比DEMO和AIDA模型高8.1%和27.0%。在AlphaFold Protein Structure Database(AFDB)中系统选取的293个人类多域蛋白质上,经SADA重新组装后的模型的平均TM-score比AlphaFold2的全链模型高出1.1%。SADA服务器参加了全球持续蛋白质结构预测竞赛CAMEO,在近三个月(2022-3-18至2022-06-11)的评测中,也获得了所有服务器(包括公开和匿名)中排名第一的成绩。进一步,在部分蛋白质复合物初步测试中,相比于AlphaFold-multimer和RoseTTAFold等算法,实验结果表明尽管SADA是面向多结构域单链建模设计,但是也表现出了组装不同链间域-域相互作用的可能和潜力。该论文通讯作者为浙江工业大学信息工程学院张贵军教授,浙江工业大学信息工程学院彭春祥博士生为第一作者、周晓根博士为共同第一作者。

图1. Bioinformatics期刊发表论文
1. 研究路线
SADA的研究路线分为五个部分:(1)根据输入的蛋白质结构域模型从构建的MPDB库中检测全链结构类似物;(2)根据检测到最好的结构类似物构建初始模型;(3)利用深度学习网络GeomNet预测残基间的距离分布;(4)基于预测的残基距离分布和多域蛋白的性质构建一个多域蛋白特定的能量函数来指导域组装;(5)通过两阶段差分进化算法来执行域组装,从初始模型优化到最终的全链模型。SADA算法流程如图2所示。

图2. SADA
算法流程图
2. 实验结果
SADA与同类域组装方法比较
为了公平地比较SADA与其它方法的性能,SADA首先在 DEMO的数据集上进行测试,该测试集包含 356 个多结构域蛋白质(其中有166 个2域蛋白、69个3域蛋白、40个超过4域的蛋白和81个不连续域蛋白)。与基于域间铰链区采样的组装方法AIDA以及基于对接的域组装方法DEMO相比,在给定测试集上SADA所组装的全链模型获得了更高的TM-score。总体而言,SADA模型的平均TM-score为0.80,分别比AIDA (0.74) 和DEMO (0.74) 高出27.0%和8.1%,几乎在所有类型的多域蛋白质上都处于领先地位,如图3所示。

图3. 在不同多域蛋白类型上SADA、DEMO和AIDA组装模型的比较结果
SADA与AlphaFold2(全链建模方法)在人类多域蛋白上预测精度比较
AlphaFold2在蛋白质结构预测上取得突破性的进展,基本解决了单结构域蛋白质预测问题,但对于多域蛋白质的预测依旧存在一定的问题。可能的两个主要原因是(1)多域蛋白的复杂性以及域间方位的准确预测比单域蛋白要难的多;(2)在PDB库中可用的同源多域蛋白相比于单域蛋白较少。因此,准确预测多结构域蛋白质域间方位仍然是一个需要关注的重要问题。论文中作者用SADA重新组装了从AFDB中随机选择的293个多域蛋白质。293个多域蛋白的选取标准如下:(1)在PDB中的天然结构中有超过90%残基已被解析;(2) 与距离预测网络GeomNet的训练集的序列相似度小于40%;(3) 经上述步骤筛选出的多域蛋白质以序列相似度30%去冗余。
按照上述规则选取的293个人类多域蛋白质上,经SADA重新组装后的模型的平均TM-score比AlphaFold2的全链模型得分高出1.1%。图4显示了AFDB中的模型(AlphaFold2计算模型)和SADA重新组装之后模型的TM-score分数比较结果。在这293个蛋白质上经SADA重新组装之后的模型有22个TM-score提升大于
0.1。特别是,对于96个TM-score小于0.9的AlphFold2模型,经SADA重新组装之后,全链模型的平均TM-score从0.66提升到了0.70,提升了6.1%。图5展示了2域蛋白1s8oA组装结构,AlphaFold2预测的全长模型的TM-score为0.72,而每个域模型的精度均高于0.95。这说明域的方向是不正确的。通过SADA对其重新组装后,全长模型的精度达到了0.97,表明SADA算法捕获到了正确的结构域方向。

图4. AlphaFold2 预测的全链模型与SADA 重组之后的全链模型TM-score比较(三角形表示AlphaFold2预测的模型的TM-score大于等于0.9,圆圈表示AlphaFold2预测的模型的TM-score小于0.9)

图5. SADA组装的全长模型和AlphaFold2全链预测的模型比较结果(2-域蛋白1s8oA)
为了让用户了解在什么情况下使用SADA能够产生更好的全长模型。在这293个人类多域蛋白上,作者分析了AlphaFold2全长模型的平均pLDDT与SADA重组模型的TM-score提升率的关系,如图6所示。从图中可以看出,比如对于平均pLDDT小于75分的AlphaFold2多域蛋白模型,他们发现,经SADA重新组装后有89%的概率能提升模型的精度。
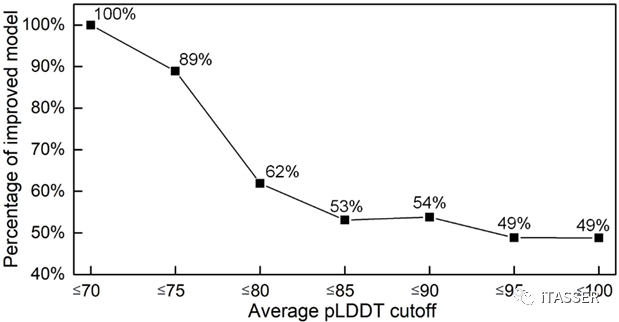
图6. AlphaFold2全长模型的平均pLDDT与
SADA重组模型的TM-score提升率的关系
CAMEO测试结果
CAMEO是全球持续蛋白质结构预测竞赛的英文缩写,由瑞士生物信息研究所和巴塞尔大学联合举办,与CASP被认为是蛋白质结构预测领域最重要的两项比赛。但不同于CASP的是,CAMEO的参赛者需要每周预测20个由世界范围内的结构生物学家最新破解出结构的蛋白质的结构,比赛的得分与排名也会每周实时更新。SADA在近三个月(2022-3-18至2022-06-11)的CAMEO评测中,也获得了所有服务器(包括公开和匿名)中排名第一的成绩,其中参赛服务器包括pureAF2_notemp和pureAF2_orig两个AlphaFold2算法服务器。

图7. SADA连续3月CAMEO测试结果
(https://www.cameo3d.org/modeling/3-months/difficulty/all/?to_date=2022-06-11)
SADA在预测复合物上的潜力
为了探索域-域之间的相互作用是否有可能拓展到链-链之间的相互作用,作者他们初步拓展了SADA来探究这一问题。在SADA的拓展版本中,每条链被当作一个虚拟的“域”,并且“域”间的链接性被忽略了。通过在8个复合物上的测试表明通过SADA这种域组装的方式是有可能探索到链与链之间的相互作用关系的,这里SADA组装的复合物的单链结构是用 AlphaFold2预测。2个代表性的例子展示在图8中。更详细的研究也将会在后续工作中开展。

图8. SADA、AlphaFold-Multimer和RoseTTAFold预测的蛋白质复合物结构比较
3. 结论
SADA是一种基于结构类似物和深度学习的蛋白质结构域组装方法,基于检测到的结构类似物利用两阶段进化算法来进行域组装,并利用深度学习预测的域间残基几何约束作为距离势能进一步辅助域组装。在356个非冗余的多域蛋白质上的测试结果表明,SADA的域组装性能要优于目前主流的域组装方法。在293个来自于AFDB的人类多域蛋白质上的组装结果表明,在大部分多域蛋白质上SADA能够捕获到更加正确的域方向,进而组装出更加准确的全长模型。通过该工作,作者他们也发现单结构域蛋白的相似性与多域蛋白(甚至是复合物)整体相似性之间存在内在相关性、同源模板和结构类似物在多结构域蛋白质全链建模中存在内在互补性的可能性,这些都将有望进一步改善模型和方法性能。
此外, SADA服务器还提供了(1)多域蛋白质的筛选功能,可以根据蛋白质长度、分辨率、域数量、R-value和序列相似度从MDPB中筛选出满足条件的多域蛋白质;(2)结构类似物检测功能,根据提供域模型的检测其全长结构类似物。
该研究得到了国家科技创新2030“新一代人工智能”重大项目、国家自然科学基金项目及浙江省自然科学基金重点项目支持。
SADA原文链接:
https://doi.org/10.1093/bioinformatics/btac553
SADA服务器链接:
http://zhanglab-bioinf.com/SADA




