单细胞转录组数据三五年前(2018)的10x技术一般来说都是3000左右的细胞数量每个样品,费用也是三万多,差不多了10块钱人民币一个细胞,那个时候基本上无需担心双细胞问题。(下面是2018的价格)
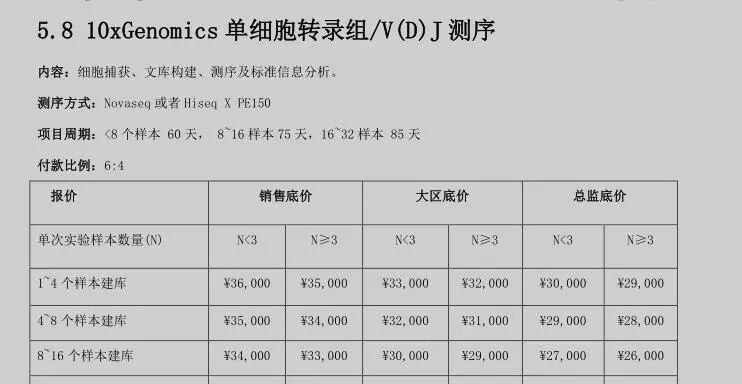
但随着10x技术的迭代,目前一般来说都可以大多8000个左右的细胞费用降低到1.7万人民币左右,相当于2块钱一个细胞。这个时候就很多人喜欢要求细胞数量越多越好,我看到很多数据集都是1.5万个细胞甚至3万个细胞的数量每个10x单细胞转录组样品,实在是让人很无语。
这样的话,我们就不得不对每次得到的10x单细胞转录组表达量矩阵进行双细胞的去除了,有一个基于Python的Scrublet工具去除双细胞速度非常快,值得推荐。
一般来说单细胞转录组测序数据走完cellranger的定量流程,每个样品就会得到3个表达量矩阵文件(barcodes.tsv.gz,matrix.mtx.gz,genes.tsv.gz或者features.tsv.gz),然后就可以走seurat流程进行单细胞降维聚类分群。但是因为要插入Python的Scrublet工具去除双细胞流程,就变得复杂了一点。
首先是Python的Scrublet工具安装
很简单的 pip install 即可,一般来说大家的服务器或者苹果电脑都是有Python环境的,也有pip这个安装器,就是需要注意一下选择合适的镜像:
pip install -i https://pypi.tuna.tsinghua.edu.cn/simple scrublet
然后写一个脚本( run_scrublet.py ):
有意思的是这个脚本名字居然还有讲究,之前命名是(scrublet.py)一直报错,后来把脚本的名字修改为 run_scrublet.py :
import sys
print(sys.path)#查看python安装位置
import os, sys
os.getcwd()
os.listdir(os.getcwd())
# 1.加载Scrublet需要的包
import scrublet as scr
import scipy.io
import matplotlib.pyplot as plt
import numpy as np
import os
import pandas as pd
# 1.读取输入文件(输入文件为10X的标准脚本)
# input_dir = '/home/bakdata/x10/jmzeng/matrix'
input_dir = sys.argv[1]
counts_matrix = scipy.io.mmread(input_dir + '/matrix.mtx.gz').T.tocsc()
out_df = pd.read_csv(input_dir + '/barcodes.tsv.gz', header = None, index_col=None, names=['barcode'])
scrub = scr.Scrublet(counts_matrix, expected_doublet_rate=0.06)
doublet_scores, predicted_doublets = scrub.scrub_doublets(min_counts=2, min_cells=3, min_gene_variability_pctl=85, n_prin_comps=30)
# doublets占比
print (scrub.detected_doublet_rate_)
out_df['doublet_scores'] = doublet_scores
out_df['predicted_doublets'] = predicted_doublets
out_df.to_csv(input_dir + '/doublet.txt', index=False,header=True)
out_df.head()
如果你有一个cellranger的定量流程的outputs文件夹,每个样品就会得到3个表达量矩阵文件(barcodes.tsv.gz,matrix.mtx.gz,genes.tsv.gz或者features.tsv.gz),很容易对单个样品跑上面的流程。
python run_scrublet.py your_out_by_cellranger
但是我们通常情况下都不会仅仅是一个样品。
批量运行这个脚本
很简单的,比如我的授课项目(PRJNA768891-ccRCC)路径下面有4个样品:
$ ls -d /home/bakdata/x10/jmzeng/2022-PRJNA768891-ccRCC/rna/outputs/matrix/*
/home/bakdata/x10/jmzeng/2022-PRJNA768891-ccRCC/rna/outputs/matrix/SRR16213611
/home/bakdata/x10/jmzeng/2022-PRJNA768891-ccRCC/rna/outputs/matrix/SRR16213612
/home/bakdata/x10/jmzeng/2022-PRJNA768891-ccRCC/rna/outputs/matrix/SRR16213613
/home/bakdata/x10/jmzeng/2022-PRJNA768891-ccRCC/rna/outputs/matrix/SRR16213614
那么,我们就批量运行这个脚本,使用下面的代码:
ls -d /home/bakdata/x10/jmzeng/2022-PRJNA768891-ccRCC/rna/outputs/matrix/* |while read id;do(nohup python run_scrublet.py $id & );done
一般来说单细胞转录组测序数据走完cellranger的定量流程,每个样品就会得到3个表达量矩阵文件(barcodes.tsv.gz,matrix.mtx.gz,genes.tsv.gz或者features.tsv.gz),但是我这个脚本并不需要基因信息,仅仅是读取表达量矩阵即可判断细胞是否是双细胞,然后读取barcodes.tsv.gz后把细胞是否是双细胞这样的信息写出来即可。
每个样品对应的文件夹里面都会输出一个文本文件 (doublet.txt ):
$ head doublet.txt
barcode,doublet_scores,predicted_doublets
AAACCCAAGCAATTCC-1,0.020104244229337306,False
AAACCCAAGCATCGAG-1,0.10305343511450382,False
AAACCCAAGCTGGTGA-1,0.05794460641399417,False
AAACCCAAGGTGGGTT-1,0.014997115939242453,False
AAACCCAAGTGTTGTC-1,0.05228981544771018,False
AAACCCACAAATGGTA-1,0.04285714285714286,False
AAACCCACAACGGCTC-1,0.007255723960012898,False
AAACCCACACAGTCAT-1,0.03284527107241788,False
AAACCCACACGCTGCA-1,0.015420424789060229,False
当然了,其实还可以出一个umap去可视化被Python的Scrublet工具预测的双细胞:
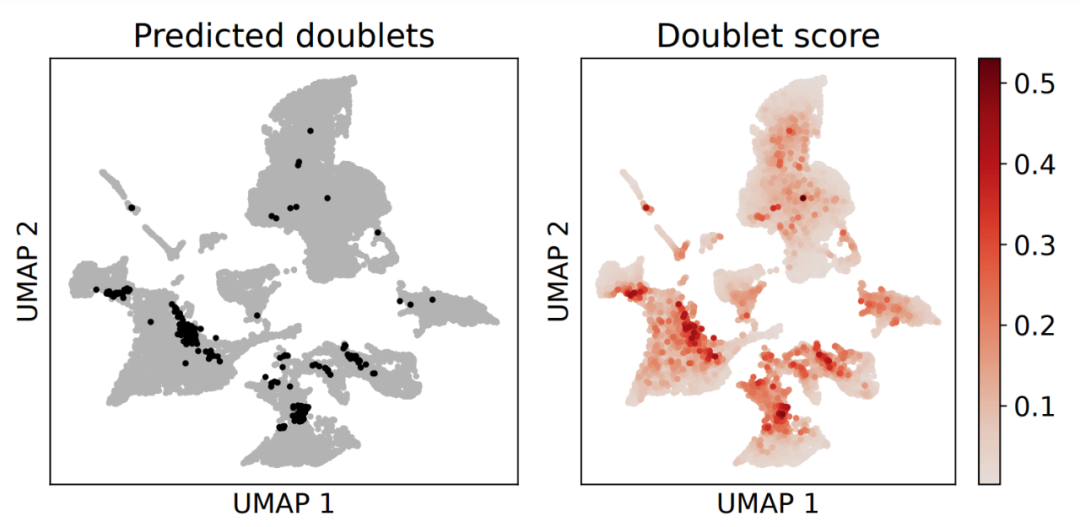
需要注意的是Python的Scrublet工具并不是帮你去除双细胞,仅仅是预测给你。后续我们在进行降维聚类分群的时候就需要读入这个文本文件 (doublet.txt ),自己进行去除双细胞操作。我的示例代码是:
###### step1:导入数据 ######
library(data.table)
dir='matrix/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
folder=file.path( dir ,pro)
print(pro)
print(folder)
print(list.files(folder))
sce=CreateSeuratObject(counts = Read10X(folder),
project = pro ,
min.cells = 5,
min.features = 500)
doub = fread(file.path( dir ,pro,'doublet.txt'))
print(table(doub$predicted_doublets))
rm_barcodes = doub$barcode[doub$predicted_doublets]
print(sce)
sce=sce[,!colnames(sce) %in% rm_barcodes]
print(sce)
return(sce)
})
names(sceList)
后面就是走走seurat流程进行单细胞降维聚类分群。这样的基础分析,也可以看基础10讲:









