二、结果
1、鉴定与进展为严重登革热相关的 8 基因组
作者搜索了截至 2019 年 8 月 1 日的 NCBI 基因表达综合 (GEO) 存储库,通过查询“登革热”获取通过阵列或高通量测序分析感染结果不同的人类登革热患者的血液基因表达的数据集。排除与研究无关的信息,由此确定了11个公开可用的数据集,这些数据集存在生物学、临床和技术异质性。利用这些数据集对365名登革热患者的血液转录组进行了分析,其中 199人的症状不严重, 166人进展为SD,其中作者将单纯性登革热 (DF) 患者分类为“非重症”,将登革出血热 (DHF) 或登革休克综合征 (DSS) 患者分类为“SD 进展者”。
接下来,作者使用MetaIntegrator在数据集上进行了迭代蒙特卡洛采样的多队列分析(图1A),以识别非重症患者和 SD 进展者之间的差异表达基因 (DEG)。 在所有迭代中发现了 25 个具有一致效应大小的显著的差异表达基因(图1B),在这25个基因表达基因中,作者执行了贪婪的前向搜索以确定最具预测性的基因集,由此得到三个上调基因和五个下调基因的 8 基因组(图1C-D)。
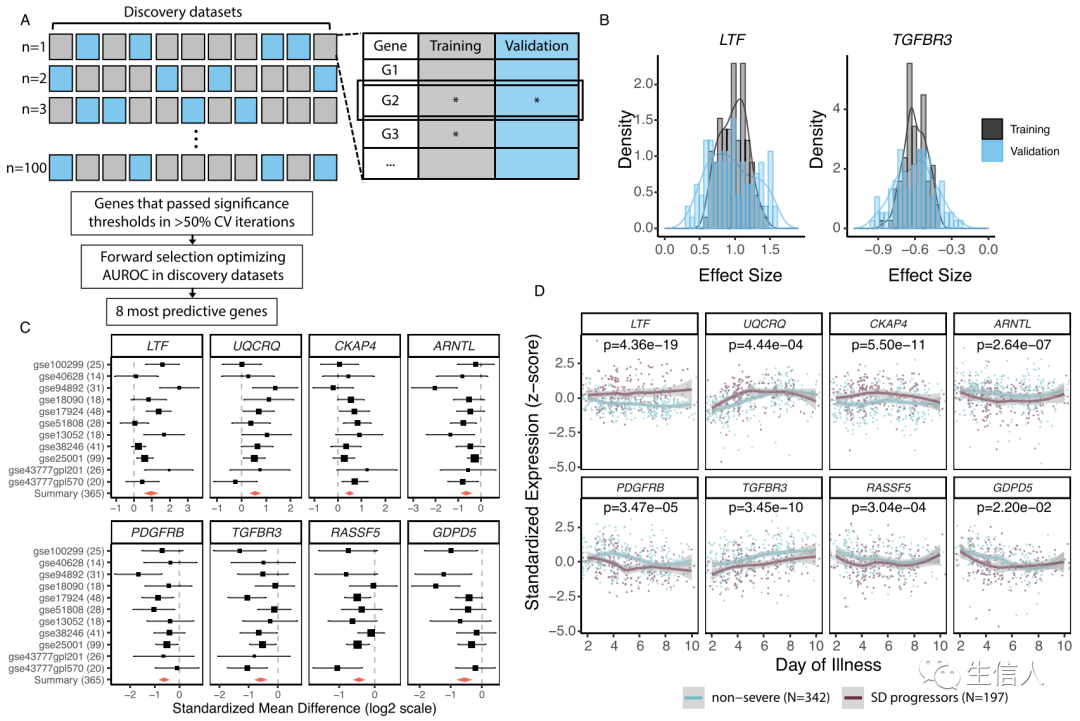
图1 多队列分析确定了八个与发展为 SD 密切相关的基因
2、构建模型以预测现有队列中的严重登革热进展
为了建立一个可推广的模型来预测 SD 进展,作者将年龄作为一个混杂变量进行了检查。此外由于许多公共数据集中缺乏样本级别的年龄信息,无法将年龄作为变量包括在内。故作者采用非线性分类器以便更好地学习年龄、基因表达和登革热严重程度之间潜在的复杂关系。
作者利用这八个基因作为特征训练了 XGBoost 梯度提升树模型。其中LTF、UQCRQ、TGFBR3和RASSF5这四个基因共同对模型预测准确度的相对贡献为72.9%(图2A),结果表明,该模型AUC= 0.891 (95% CI 0.706-1),在Youden阈值下,具有 89.2% (95% CI 84.1–93.8) 的敏感性和 81% (95% CI 75.0–86.7) 的特异性(图2B),这表明基于8 基因组 的XGBoost 模型具有更高的泛化性。但基于8 基因组 的XGBoost 模型应用于不同于模型训练的公共数据集时,其效果不佳,故作者同时评估了基于20 基因组以及 8 基因组 XGBoost 模型。
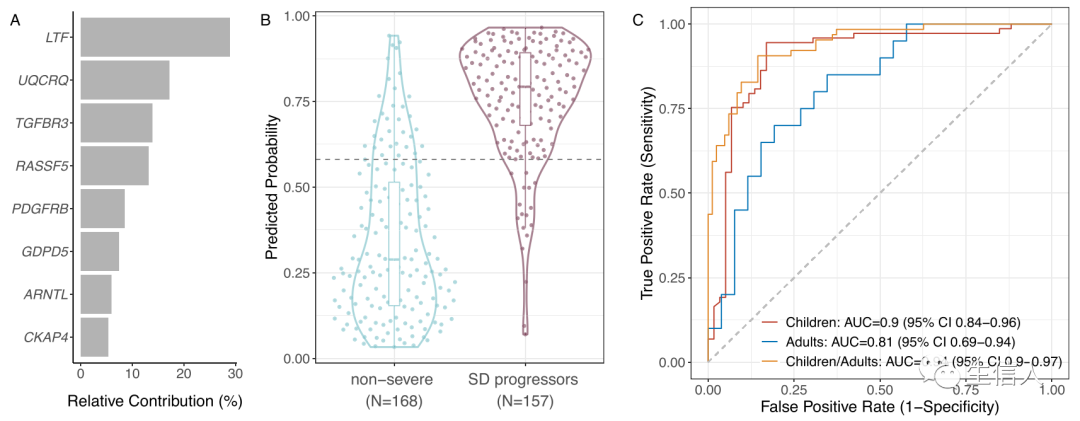
图2:基于 8 基因 XGBoost 的模型预测公共数据集中的 SD 进展。
3、在登革热患者的前瞻性队列中独立验证和与临床警告信号进行比较
在收集独立性数据阶段,作者前瞻性地在哥伦比亚卡利和布卡拉曼加招募了 377 名 DENV 感染患者,出现 SD 的患者被排除在外。
首先作者分析了出现临床警告信号预测是SD进展时的准确性。作者在就诊时(即在进展为 SD 之前)收集全血样本,并在整个感染过程中跟踪患者(图3A)。在就诊时临床警告信号预测是SD进展的敏感性为77.3% (95% CI 58.3-94.1),特异性为39.7% (95% CI 34.7-44.9)(图3C)。在成人中,临床警告信号预测是 SD 进展的敏感性和特异性分别为 66.7% 和 45.2%。在儿童中,临床警告信号预测是 SD 进展的敏感性和特异性分别为90.0%和37.1%。
总的来说,出现临床警告信号预测是SD进展的(阳性预测值)PPV为7.4% (95% CI 4.3-10.9), NPV(阴性预测值)为96.6% (95% CI 93.3-99.3),需要25.4例NNP,其中NNP 被定义为需要检查以准确预测一名患者将进展为 SD 的登革热患者的数量。
接下来,作者将基于8基因组的XGBoost模型应用到该独立前瞻性队列中,该模型预测随后进展为 SD的AUC 为 0.844(95% CI 0.749-0.938),在 Youden 阈值下,其敏感性为 86.4%(95% CI 68.2-100.0),特异性为 79.7%(95% CI 75.5-83.9),阳性和阴性似然比为 4.3(95% CI 3.2–5.5) 和 0.2 (95% CI 0.01–0.4)(图3C)。与临床警告信号相比,8 基因模型的 PPV 和NPV显著更高,NNP 减少了 80%。8 基因模型的年龄差异无统计学意义(DeLong p = 0.19),相比之下,之前的 20 个基因组在成人中的表现比在儿童中的表现更差(DeLong p = 0.0026)。因此,与临床警告信号相比,8 基因 XGBoost 模型改善了两个年龄组的预测,并且比 20 基因组更具有普遍性。
此外,鉴于登革热进展快速的性质,作者检查了8 基因模型在疾病过程中的时间表现(图3D-F)。8 基因模型可预测整个疾病过程中收集的样本的随后的 SD,此外,对于SD患者,8 基因模型其进展为 SD 的前三天预测也是准确的。
最后,作者利用三个公共数据集按照1997年WHO标准分析了8基因模型的性能,模型性能与 1997 年标准相当(AUC = 0.842, 95% CI 0.716-0.968)。
总的来说,这些结果证明了 8 基因模型的早期预后能力和普遍性,适用于在大型、独立、前瞻性登记的队列中收集的样本。
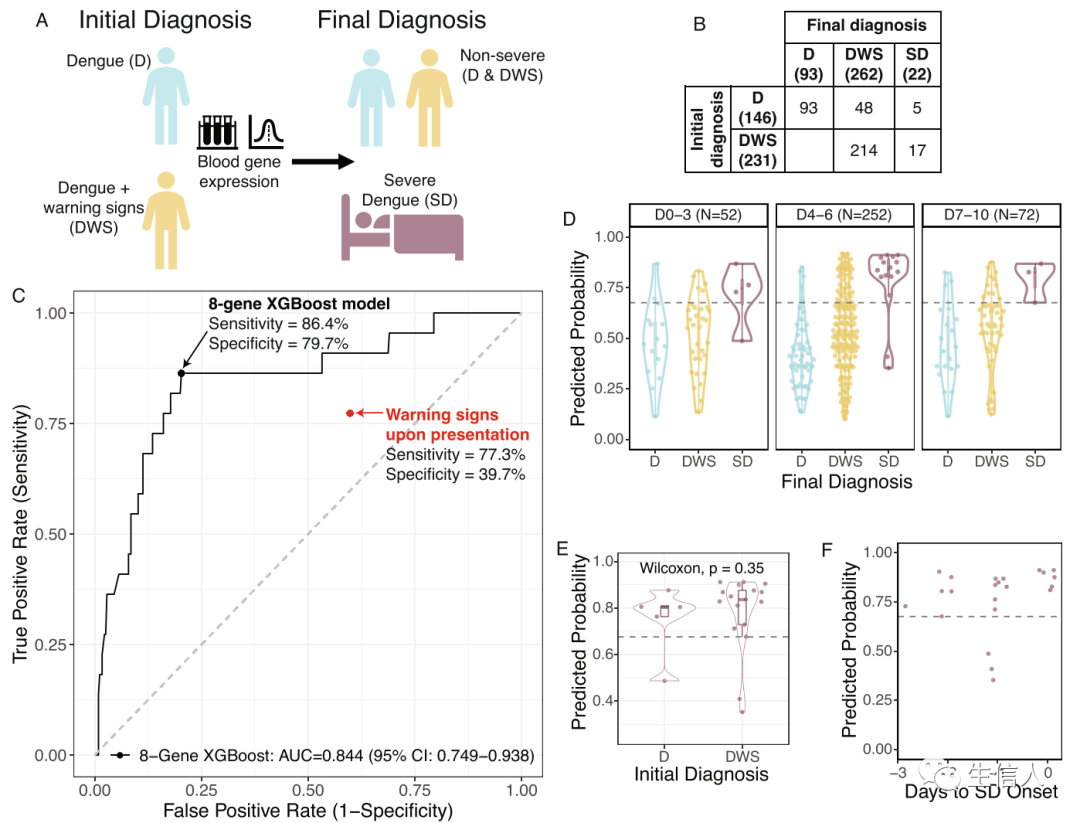
图3:基于 8 基因组的 XGBoost 模型在独立的前瞻性登革热队列中预测进展为 SD的效果
4、临床特征与 8 基因组模型预测概率的关联性
作者接下来检查了 8 基因组模型预测与相关临床特征之间的关系。8 基因组模型预测的概率在先前接触过 DENV 的患者中显著高于未接触过的患者,但该模型能准确区分了原发性或继发性感染的 SD 进展者。此外,8 基因模型预测与体液积聚呈正相关,但与呕吐、出血、腹痛或肝肿大无关,与峰值丙氨酸转氨酶(ALT)和天冬氨酸转氨酶(AST)呈显著中度正相关,与血小板最低点呈中度负相关。
5、8 基因组模型对其他病毒感染的普遍性
作者评估了 8 基因组模型是否也可以预测其他病毒感染的严重程度。作者确定了四个独立的队列,包括 336名感染 SARS-CoV-2、基孔肯雅热、流感或呼吸道合胞病毒 (RSV) 的患者。结果表明,8 基因组模型能区分轻度/中度感染与严重感染 SARS-CoV-2、流感和 RSV,但不能区分基孔肯雅热,说明其在其他病毒感染中可能具有一定的区分能力。







