导读
目前,世界各地的病理实验室对患者标本大多进行常规的福尔马林固定和石蜡包埋(Formalin Fixation and Paraffin Embedding, FFPE)处理。FFPE保留了组织形态,可用于临床诊断的免疫组化分析。但与新鲜冷冻样本相比,FFPE样本来源的DNA质量和数量均有负面影响,并可能引入人工突变,严重限制了对FFPE样本的突变特征分析。很多大型医院的病理科都拥有数以万计的FFPE样本,因此实现FFPE样本的精确基因组分析,将有助于释放这些海量样本巨大的研究和应用潜力。美国赫尔辛基大学研究团队在Nature Communications发表了一篇题为“The mutational signatures of formalin fixation on the human genome”的研究论文。研究团队提出了一种准确分析癌症活检FFPE样本中基因组学数据的方法,使用机器学习方法校正了受损DNA,并揭示肿瘤样本中真正的突变过程。该方法能够对FFPE样本进行稳健的突变特征分析,从而为使用FFPE-WGS数据进行临床应用铺平了道路,有助于在数百万现有癌症储存样本中释放巨大的医学价值。
文章发表在Nature Communications
当福尔马林固定时间较长时,由福尔马林损伤引起的突变数量会增加。研究团队发现,FFPE样本中C>T突变量与福尔马林固定时间呈正相关,证实C>T突变是福尔马林诱导的人工突变。为了获得FFPE引入的突变特征,研究团队使用t-SNE聚类来选择有代表性的样本,以排除可能的异常值,并执行了一个迭代过。为了验证计算得到的FFPE样本中突变特征的稳健性,研究团队从不同研究中进行了独立的特征推断,发现了几乎相同的突变模式。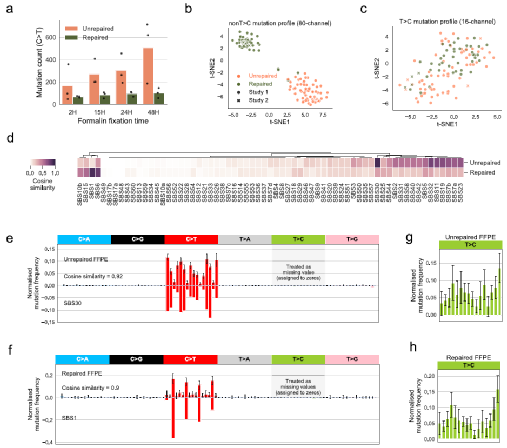
图1. 福尔马林处理后的突变特征,来源:Nature Communications接下来,研究团队设计并实现了一种名为FFPEsig的算法,用于校正FFPE处理诱导的突变。FFPEsig算法将给定FFPE样本中观察到的聚合突变目录分解为FFPE-伪信号(噪声)和真实的生物突变信号,并在模拟中省略了T>C突变,以匹配真实样本。在这一设置下,FFPEsig以约98%的精度成功推断出生物突变目录。研究人员随后将FFPEsig引用于泛癌全基因组分析项目(Pan-Cancer Analysis of Whole Genomes, PCAWG)中的所有样本(n=2780)。结果显示,FFPEsig对未修复-FFPE和修复-FFPE样本的平均校正精度分别为0.90和0.87。虽然FFPEsig在不同癌症类型中的校正精度有差异,但整体水平仍然很高,其中在皮肤黑色素瘤中的准确率最高(约0.98),其次是膀胱移行细胞癌(约0.96)和肺鳞癌(约0.96)。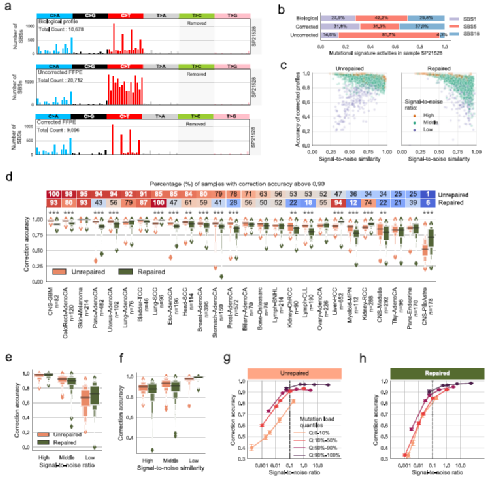
图2. FFPEsig算法及使用FFPEsig进行校正,来源:Nature Communications
在上述分析中,由于来源不明,研究团队选择忽略了T>C突变。为分析去除T>C突变对FFPEsig校正的影响,研究人员对来自2726个PCAWG基因组29个突变特征的10312个突变进行了重组(另外54个基因组由于重建精度较低或样本量过小而被排除)。分析数据显示,29个特征中有24个实现良好的校正。在校正效果较差的5个基因中,有4个基因T>C突变率较高,分别为SBS7d、12、16和17a。此外,SBS6突变存在较高的不一致率,主要在非霍奇金淋巴瘤中被检测到。总的来说,去除T>C突变对大多数病例的FFPEsig校正分析影响很小。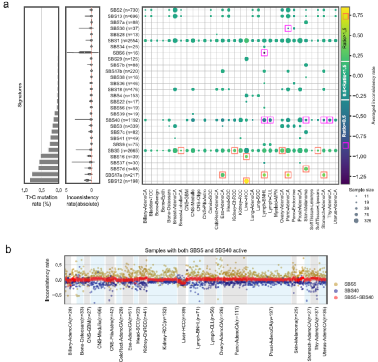
图3. T>C突变对FFPEsig校正性能的影响,图片来源:Nature Communications
综上所述,研究团队从FFPE样本中提取了福尔马林暴露导致的全基因组突变特征,并设计了FFPEsig算法。该算法可将样本突变谱中去除FFPE处理诱导的突变。研究发现,修复和未修复的福尔马林样本突变主要为C>T突变,这一突变特征在独立的实验研究中是一致的。因此,FFPEsig算法的应用将使世界各地大量FFPE样本的突变特征获得稳健的深入探索。文章第一作者Qingli Guo博士表示:“福尔马林会对DNA造成严重损伤,对分析保存组织样本中的癌症基因组提出了不可避免的挑战。由于DNA质量差,使得这一宝贵资源目前尚未用于分子诊断。我们的结果表明,如果没有噪声校正,通常会错过近一半的癌症过程,而使用FFPEsig,其中90%以上的过程则可被准确预测。”Qingli Guo博士补充道:“该研究发现能够从保存在室温下数十年的肿瘤活检样本中表征临床相关特征,并可对福尔马林如何影响癌症基因组进行更加深入的了解。”虽然如此,研究人员还指出,该方法目前并不能完全去除FFPE样本中出现的批次效应的噪音,而且该工具的性能因癌症类型而异,因此必须谨慎分析所有发现。同时,研究人员也有兴趣在未来进一步将该方法应用于更广泛的存档样本研究。参考文献:
1. Guo, Q., Lakatos, E., Bakir, I.A. et al. The mutational signatures of formalin fixation on the human genome. Nat Commun 13, 4487 (2022).
2. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
3. Chakravarty, D. & Solit, D. B. Clinical cancer genomic profiling. Nat. Rev. Genet (2021).
往期精品(点击图片直达文字对应教程)
机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集