传统的蛋白质序列设计方法,如Rosetta中FastDesign,是利用rotamer library对氨基酸进行采样,在势能面上采用蒙特卡洛模拟不断循环迭代得到能量最低的序列。继深度学习在蛋白质结构预测领域大放异彩之后,在蛋白质序列设计领域也有了出色的表现。David Baker组今年6月在Biorxiv上发表了Robust deep learning–based protein sequence design using ProteinMPNN,到9月15号被Science接收。该方法广泛适用于单体、环状低聚物、蛋白质纳米颗粒和蛋白质-蛋白质界面的设计,并且进行了实验验证。相对于传统的Rosetta Fastdesign,MPNN计算速度更快,设计序列的准确性也更高[1]。ProteinMPNN基于structured-transformer [2],采用了3层encoder,3层decoder和128隐层的自回归模型架构。基线模型的sequence recovery大概在41.2%左右,改进后达到了50%以上。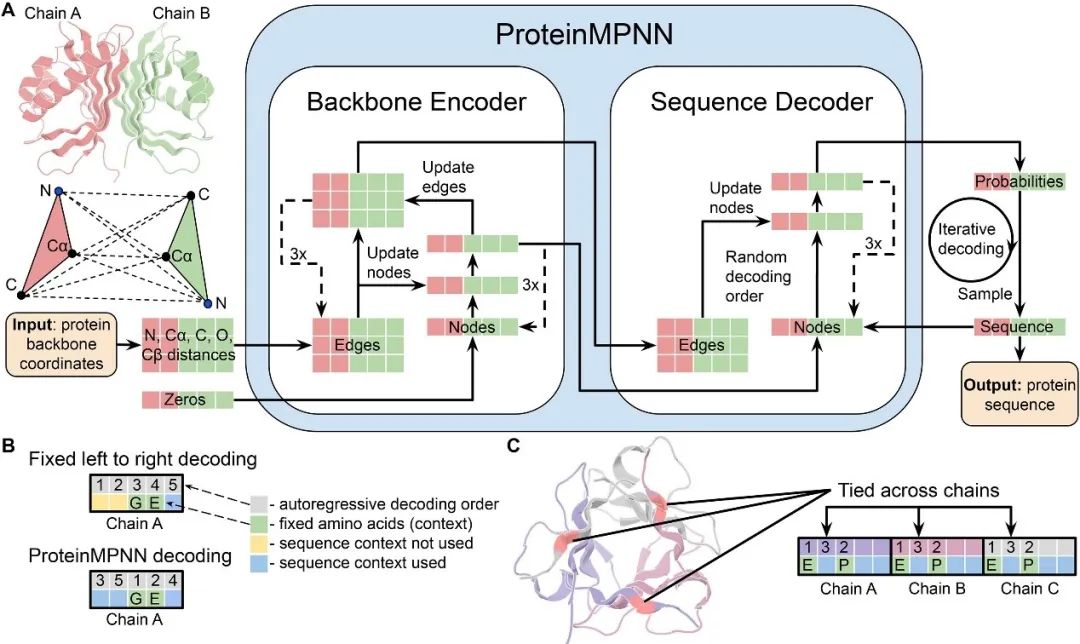
改进的方向主要有以下几点:
1.输入信息:除骨架坐标外增加了关于N,
Cα, C, O,以及虚拟Cβ原子(表示主链二面角特征)的距离信息,模型表现提升到49.0%。这表明使用pair-wise等有朝向性的特征能更好地捕捉到蛋白内部的相互作用信息;
2.在encoder中引入了边的更新机制,增加了Cα的邻近神经网络数目(32-48个时模型表现达到饱和)信息的交互,让模型表现小幅提升至50.5%。与蛋白质结构预测任务不同,序列设计任务中氨基酸的优化更多地取决于即时的蛋白质环境。
3.为了使模型广泛应用于单链/多链等设计任务,MPNN对decoder中解码顺序进行随机采样,替代了传统的语言模型固定的N->C端解码,模型提升0.2%。虽然性能提升不大,但是这种机制可以在蛋白序列设计中引入已知的motif或受体等固定信息。
4.为了让模型对多链顺序等变,模型新增了相对位置的编码信息与链号标记信息;在同源多聚体的任务中,同一位点的氨基酸可以被偶联被同时解码,从而完成对称性或multi-state等相关的设计任务。
5.在训练时,对主链引入高斯噪音,增强模型的泛化能力,噪音的引入可以让模型学习到的只是更准确地映射整体的拓扑结构而不是过度精细的局部环境。——性能评估——
在402个单体骨架测试集上使用Rosetta PackRotamersMover 和ProteinMPNN 设计序列, ProteinMPNN 的平均序列恢复率为 52.4%,而 Rosetta 为 32.9%,且耗时远小于Rosetta(单个 CPU 上处理100 个残基1.2 s VS 258.8 s)。
Average Cβ distance
for 8 closest neighbors表示包埋程度,左侧为疏水核心层,序列恢复率达到90-95%, 右侧为界面层,序列恢复度仅有35%(图2.A),这表明模型能从主链特征学到了表面残基和内核残基的相对概念,由于表面的残基由于缺乏邻居信息,因此多样性比内核层高。大量事实也表明:内核层残基的序列十分保守,突变容易导致蛋白稳定性下降或不表达,而大量的表面疏水残基可以被重新设计或赋予新的功能。
Violin plot展示了ProteinMPNN在690 个单体、732 个同聚体、98 个异聚体测试集上的sequence recovery,中位序列回收率分别为单体 52%,同聚体 55%,异聚体和界面残基 51%,同聚体 53%,异聚体 51%(图2. B)。

图2 ProteinMPNN序列恢复率与Rosetta相比及在三个测试集中表现
并且,对ProteinMPNN设计出的序列进行结构预测(AlphaFold),相比天然蛋白预测准确度提升。
天然蛋白质sequence recovery只是一个评价基准,序列设计方法的真正价值仍体现在实验测试中。研究者基于之前失败的设计,保持原有的主链骨架,用ProteinMPNN重新进行设计,在大肠杆菌中进行表达,并进行了结构和生化表征。
团队首先表达了之前发表的Hallucination protein(和天然蛋白无关的“幻想”蛋白,基于AlphaFold构建的骨架及采用Markov Chain Monte Carlo生成的序列),发现大部分都不可溶。而用MPNN对幻想骨架进行序列生成,表达出的蛋白质大部分都是可溶的(96条设计序列,73条可溶)。并且热稳定性好,能维持寡聚状态。
对一条序列解析晶体结构,该α-β蛋白(包含 5 个 β 链和 4 个 α 螺旋)晶体结构与设计结构高度吻合,内核侧链电子云密度几乎完全符合,准确度极高。
因此,ProteinMPNN 可以稳定而准确地设计单体和环状低聚物的序列。
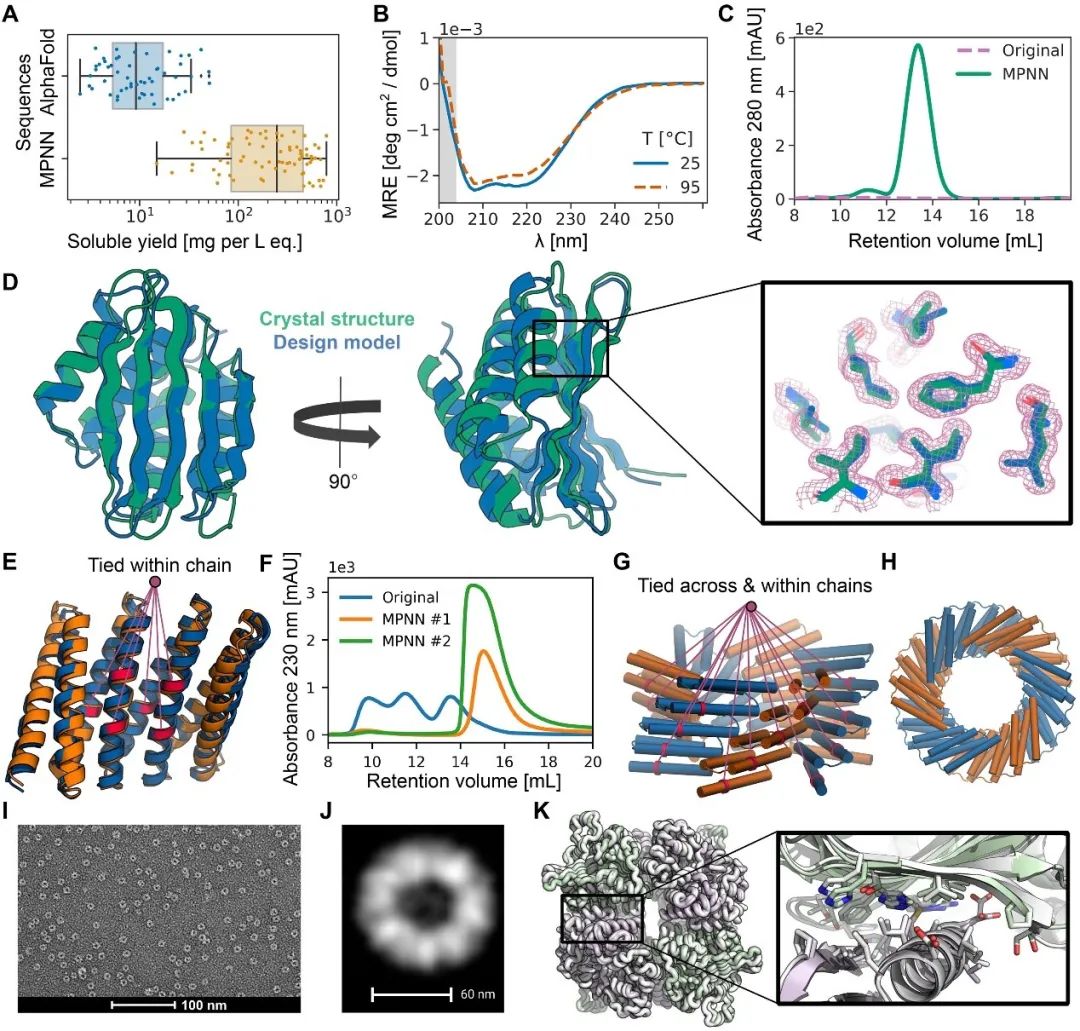
图3 A. 两种序列设计方法生成蛋白的溶解度数据 B. 圆二色谱表征热稳定性 C. 尺寸排阻色谱上的停留时间 D. 通过X-ray解析了ProteinMPNN 单体设计结构(8CYK)重复组装的多体蛋白
对C 5 /C 6环状低聚物进行实验表征,Rosetta设计的序列仅有40%可溶,且无 SEC-MALS 验证的正确寡聚状态,而ProteinMPNN 设计的集合中,88%(总共 18 个)是可溶的,27.7%具有正确的寡聚状态。结构由negative stain EM实验解析,图像均值与设计模型高度一致。
图4 I和J:电镜下的负染实验,表达蛋白的形态结构与设计基本一致ProteinMPNN最后挑战了之前Rosetta设计失败的例子:polyproline II helix motifs 与 SH3 domains识别的复合物结构。SH3-binding motif (PPPRPPK)富含脯氨酸,但在自然界十分罕见。
首先使用RIFDOCK将高度稳定的helix scaffold对接到复合物的表面,并使用RosettaRemodel将motif和scaffold进行重组和Rosetta
layer设计,但实验未表现出高结合力。而ProteinMPNN对主链进行重新设计,设计序列中两个天冬酰胺侧链和表面相互作用,实验检测出高亲和力,而点突变为天冬氨酸后无亲和力。设计结果表明MPNN可以对蛋白packing进行纠错,并产生更稳定的相互作用,也验证MPNN设计的正确性和准确性。
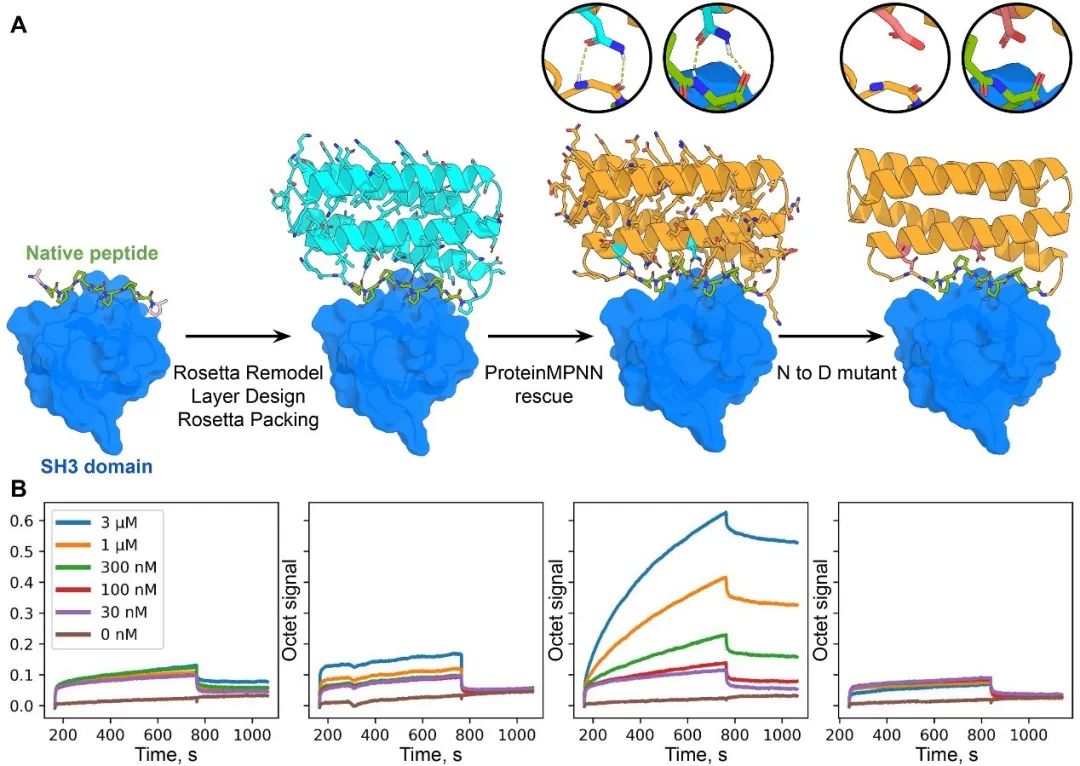
图5 B图使用的是生物膜层干涉技术biolayer
interferometry,验证了设计复合物的亲和力
ProteinMPNN相比于基于物理方法的Rosetta,不仅计算速度大大加快,sequence recovery也极高(52.4% VS 32.9%),并且解救了之前Rosetta设计失败的案例,在单体,多聚物,界面设计领域都有成功的应用。蛋白质序列设计无法成功的因素有很多:无法折叠,亲和力低,不可溶,稳定性低等等。遍历所有的构像空间不可实现,基于物理的 Rosetta在限制,描述中会有ambiguity。所以用Rosetta设计序列后还需要进一步采用计算验证(如ab initio folding,
MD),或加入人为经验的调整。而ProteinMPNN的学习方法虽然不透明,但完全基于结构和序列,实现了一步到位。它展现出的逻辑和认知相符:比如蛋白质核心的残基会受到更多相邻残基的影响,对于蛋白质正常折叠而言至关重要,进化中不会轻易突变;而表面上的残基受相邻残基的影响小,可以引入更多变化。
对蛋白质设计的任务本身而言,之后或许可以采用ProteinMPNN代替FastDesign,packing等步骤,但目前看来MPNN仍需要一个比较靠谱的初始化主链,或许在不久的将来,人们可能会更多倾向于讨论如何生产高可设计性的主链生成。MPNN的构架可能也会为其它蛋白质相关任务提供新的思路。
Github code已开源,地址https://github.com/dauparas
[1] Dauparas, J. et al. Robust deep
learning–based protein sequence design using ProteinMPNN. Science
. (2022) doi:10.1126/SCIENCE.ADD2187.[2] Ingraham, J., Garg, V. K., Barzilay, R.
& Jaakkola, T. Generative models for graph-based protein design.
点击左下角的"阅读原文"即可查看原文章。
本文为GoDesign原创编译,如需转载,请在公众号后台留言。









