基因推断(genotype imputation)近十几年来已经广泛应用于遗传分析中。通过有效借助推断所用的参照基因库(reference panel)涵盖的连锁不平衡(linkage disequilibrium)信息,基因推断方法例如MaCH/mimimac/IMPUTE/Beagle可以成功推断出大量没有被起始芯片包括的位点。图一 (出自Li et al 2009文献) 用图示阐述了基因推断的概念。其中研究或者目标样本(每个小图上方的Study sample)未做基因推断之前通常只有芯片涵盖的约百万位点的信息。随着遗传研究样本量的增加,推断所用的参照基因库也日益完善,从最初的只有几百人,三百万位点的HapMap2 参照库发展到当今广泛应用的近十万人,逾三亿位点的TOPMed reference panel。参照基因库的飞速增长也使得研究人员可以在目标样本中推断多达上亿位点的基因分型。但是这些位点大多都是低频的(low frequency)或罕见的(rare),通常在人群中的次等位基因频率(minor allele frequency,MAF)低于5%。相对于常见位点 (common variants),这些上亿的非常见(uncommon)位点更难推断。首先,绝对推断质量(imputation quality)偏低:上亿的非常见位点中,通常只有远低于半数的能符合质检进入下游分析。质检因而尤为重要。当今业界质检是基于估算的质控统计量。现有最先进的质控统计量由进行基因推断的软件直接提供,例如Minimac 4 的Rsq,IMPUTE5的INFO,以及Beagle 5的DR2。遗憾的是,现有的质控统计量 对非常见位点表现得不尽人意。这些估计量虽在具体的算法上稍有不同,但它们之间的相关性非常强,对于非常见变异都不能有很好的表现。遗传学家们早在十年前就已注意到这个问题,但一直以来都没有提出更好的估计方法来代替传统的统计量。图二a是作者在实验中所得的质量真实值(true R2)与估计值Rsq的比较,我们可以清楚看到很多位点偏离45度线,尤其是左上角的大量位点:实际高质量但被质控统计量低估,因而不能参与下游的分析,白白浪费了推断出来的这些高质量位点的信息。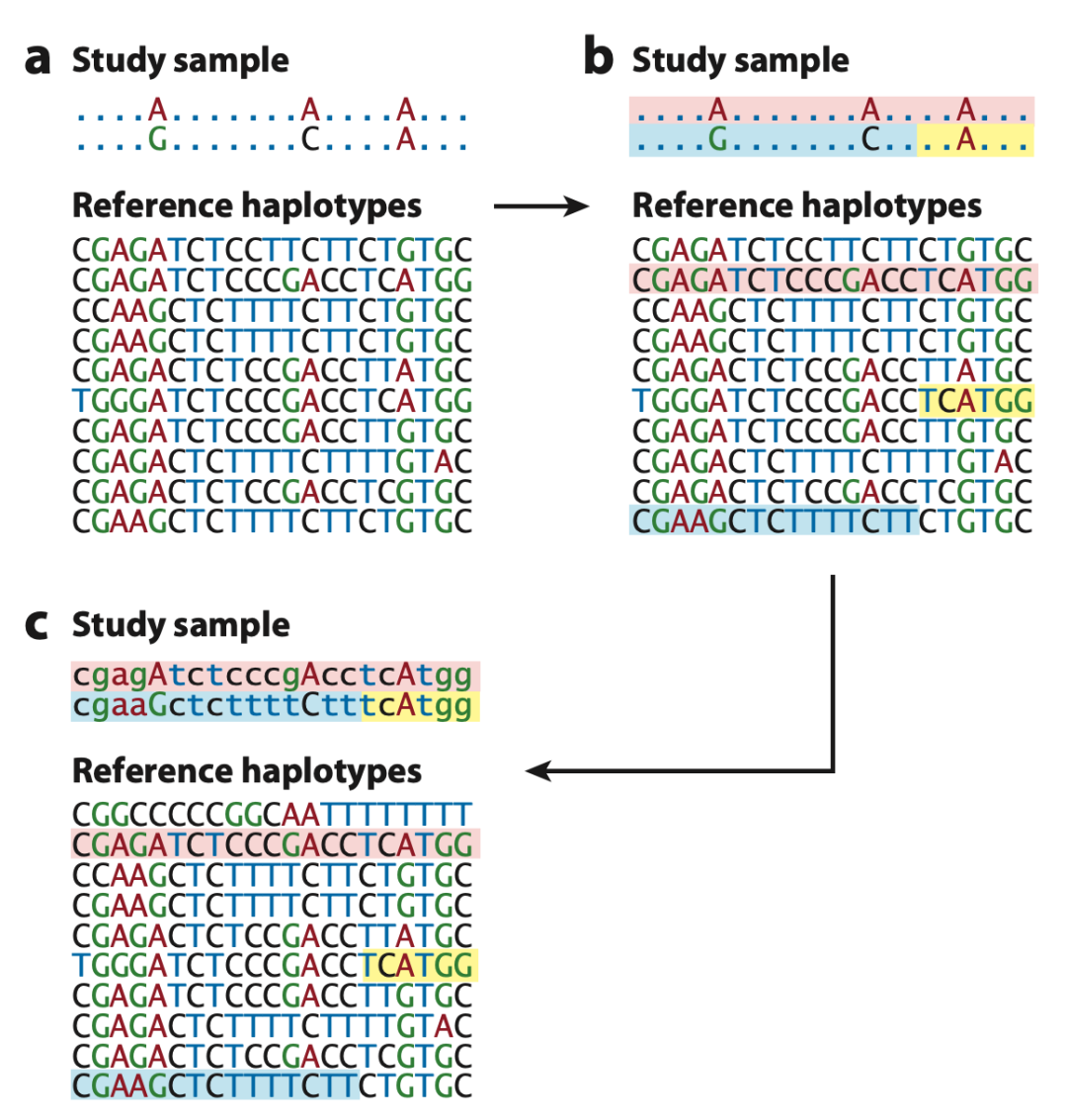

图二:a. 基因推断质量真实值和传统估计值Rsq的比较。b. 基因推断质量真实值和新方法MagicalRsq估计值的比较。
2022年10月4日,来自北卡罗来纳大学教堂山分校的遗传统计学家李蕴实验室和意大利Eurac研究中心的Christian Fuchsberger在American Journal of Human Genetics中发表了题为MagicalRsq: Machine-learning-based genotype imputation quality calibration的文章,提出了基于机器学习方法的新的基因推断的质控统计量。作者的研究结果显示,新方法MagicalRsq可将估计量与其真实值的平方相关系数(squared Pearson correlation)提高8% - 92%,并可挽救高达几百万的位点使其参与下游分析中。如图二b所示,相比于Rsq,大量位点的MagicalRsq与真实值集中于45度线附近,表明MagicalRsq能够更好地估计真实值。
MagicalRsq的模型构建基于训练数据集。在训练数据集中,每个位点是一个数据点。我们需要有上万位点的真实质量信息作为模型的预测对象。这个要求在很多研究中都很容易达成:比如当目标样本有多个芯片提供的基因分析数据,而只有其中一个芯片的数据用于基因推断;再比如当目标样本既有芯片又有测序技术提供的基因分型,同样如果只有芯片数据用于基因推断的话。以上的例子中,其他芯片或测序提供的、但没有用于基因推断的位点都可以算出真实质量 -- 通过比较推断的基因型和芯片/测序测出的真实基因型。解决了模型的预测对象问题,我们来介绍一下MagicalRsq用到的自变量。由于MagicalRsq模型中,每个数据点是一个位点,所以模型的预测对象和所有的自变量都是针对每个位点的。MagicalRsq考虑的自变量,也即是位点的样本特征,包括基因推断所得到的概括统计量(summary statistics)以及额外的群体遗传学(population genetics)概括统计量。其中基因推断的概括统计量包括次等位基因频率的估计值(estimated MAF)以及现有软件的质量估计值(Rsq),是与目标人群直接相关的。而群体遗传学概括统计量,涵盖了基因频率,自然选择等多个方面,是每个位点独立于目标人群的额外特性。基于这些特征,MagicalRsq通过构建梯度提升决策树(XGBoost)模型进行训练来预测真实质量信息。模型训练好之后便可直接利用在目标测试数据上:通过在新的目标人群中带入位点特征值,从而计算MagicalRsq。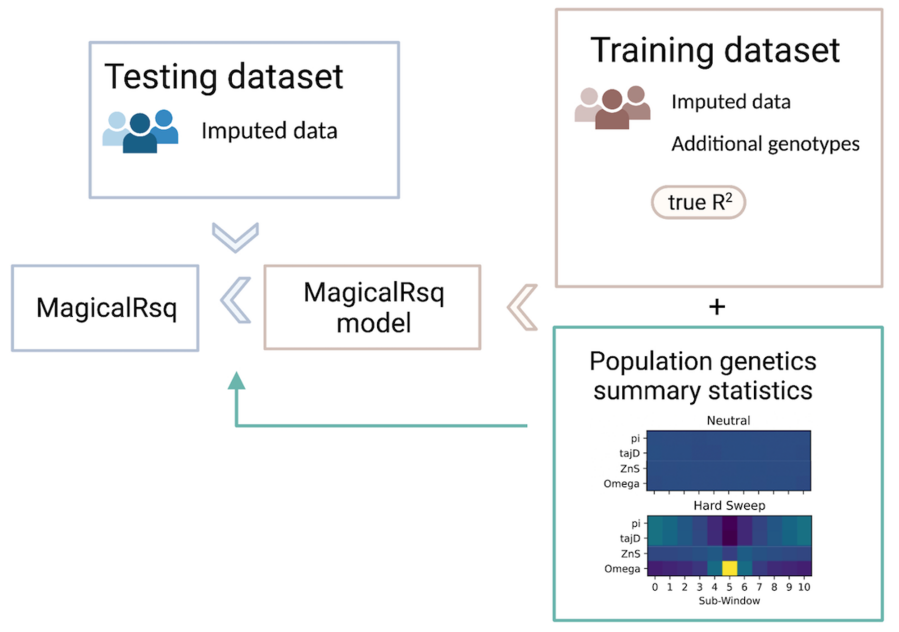
为了系统性全方位地比较MagicalRsq与传统Rsq,作者考虑了两大类型下的多种实际应用情形。第一类是所有的目标人群都有额外位点的基因分型数据,例如其他未用于基因推断的芯片信息,候选基因测序(candidate gene sequencing)信息,或者全外显子组测序(whole exome sequencing,WES)信息,第二类则进一步考虑了实际研究中更常见的只有一部分目标人群有全基因组测序(whole genome sequencing, WGS)
或部分额外的基因分型数据。研究结果表明,在第一种情形下,即使仅有一万个位点,训练所得到的模型也足以提高原本Rsq的表现,挽救几万至几十万的位点用于下游分析。而且上述模型不受训练位点所属区域的影响。比如,训练位点可以来自某一个随机选出的20MB的区域。再比如,位于外显子的位点所训练出的模型,可以应用于位于基因组内非外显子区域的位点。对于第二种情形,作者在三个不同种族群体(欧洲裔,非洲裔和南亚裔)上都进行了模型测试,其结果表明MagicalRsq对不同种族群体都有较为优异的表现,证明了MagicalRsq方法可广泛应用于各种族人群。出于对计算成本的考虑,作者系统测试了可靠MagicalRsq模型训练所需的位点数。作者尝试了用一万至一百万位点进行模型训练,并将所得模型应用于独立测试数据集去评估其表现。其结果显示,即使只用一万个位点,也可得到比常用Rsq更好的质控统计量,且模型不受所用于训练的具体位点的影响。虽然用更多位点来进行训练所得估计量更为准确,但综合考虑计算成本,其边际效益呈现递减趋势,因此作者建议用一万至一百万的位点训练模型即可。其中十万位点模型的计算时间平均在6-30分钟。在对于MagicalRsq的实际应用上,作者举了BRCA1错义突变(missense variant) rs28897686 (chr17:43091783:C:T, hg38, NM_007294.4:c.3748G>A, p.Glu1250Lys) 的例子来说明此方法可挽救一些在临床研究中起到重要作用的位点,并系统性的比较了MagicalRsq 和Rsq在挽救ClinVar数据库中潜在重要外显子位点的能力。其结果显示,15个Rsq错失的位点可被MagicalRsq挽救,而反过来,没有一个MagicalRsq 错失的位点可由Rsq挽救。这一结果虽间接却有力地表明了MagicalRsq在关联性分析及后续临床应用中的优势。MagicalRsq的模型训练及应用所需代码已公开发布于Github 网站https://github.com/quansun98/MagicalRsq,在此网站上作者还发布了用于模型训练的群体遗传学概括统计量。作者表示,研究人员可灵活根据自己所需有选择性地使用这些统计量,也可以自选其他的统计量。总结来说,新开发的MagicalRsq方法显示出其在各种实际情况中优于现有统计量Rsq的优势,并有助于下游分析和应用。这一方法的提出,是机器学习在遗传学领域的一个新的应用,填补了近十年来悬而未决的基因推断质量问题的空白,为之后该方向的发展提供了新的思路。原文链接:
https://doi.org/10.1016/j.ajhg.2022.09.009

【非原创文章】本文著作权归文章作者所有,欢迎个人转发分享,未经作者的允许禁止转载,作者拥有所有法定权利,违者必究。










