单细胞多组学分析在生物学研究中的普及,促进了人们对细胞异质性和亚群体的理解。特别是通过测序对转录组和表位进行细胞索引(CITE-seq)方案的可用性不断提高,极大地促进了相关研究进展。CITE-seq是一种单细胞多组学技术,能够同时分析RNA基因表达和细胞表面蛋白,具有发现单模态单细胞RNA测序(scRNA-seq)所遗漏的细胞异质性的潜力,目前已广泛应用于生物医学研究,特别是免疫相关疾病和其他疾病(如流感和COVID-19)。
CITE-seq分析的一个挑战是需整合多个CITE-seq和scRNA-seq数据集,数据集成增加了信息内容,同时也加剧了计算困难。此外,相较scRNA-seq数据,CITE-seq数据的生成成本也很高。对此,一个潜在的解决方案是了解RNA和蛋白质之间的关系,从大型参考数据集中借用信息,然后对scRNA-seq数据进行蛋白质预测。Seurat 4和TotalVI都已被引入来实现这一功能,但其计算成本十分昂贵,且都存在局限性。
近日,美国宾夕法尼亚大学研究团队在Nature Machine Intelligence上发表了题为“A multi-use deep learning method for CITE-seq and single-cell RNA-seq data integration with cell surface protein prediction and imputation”的文章。研究团队开发了一种多用途的深度学习方法——sciPENN,支持CITE-seq和scRNA-seq数据整合,能够预测、插补scRNA-seq、CITE-seq蛋白质表达,量化不确定性以及实现从CITE-seq到scRNA-seq的细胞类型标记转移。跨多个数据集的综合评估表明,sciPENN优于当前同类其他方法。
文章发表在Nature Machine Intelligence上
sciPENN的模型架构如图1所示,其总体目标是从一个或多个CITE-seq参考数据集中学习。当CITE-seq参考数据不完全重叠时,sciPENN可以对每个参考数据集的缺失蛋白质进行估算。在CITE-seq参考数据中学习后,sciPENN能够预测scRNA-seq查询数据集的所有蛋白质,并将多个数据集整合到一个共同的嵌入空间中。sciPENN可以估计蛋白的平均表达量,量化估算的不确定性,并选择性将细胞类型标签从CITE-seq参考数据转移到scRNA-seq查询数据中。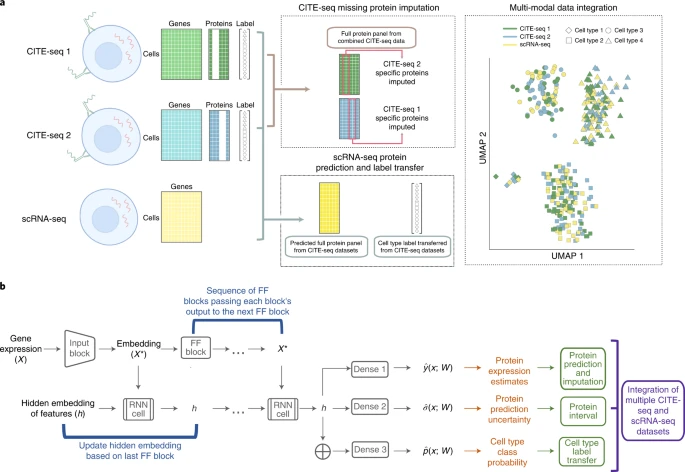
研究团队使用Seurat 4文章中报告的161,764个人类外周血单核细胞数据集(PBMC)进行分析,其包含224种蛋白质。对于测试集,使用了粘膜相关淋巴组织数据集(MALT),其包含由10x Genomics生成的8,412个细胞。在MALT数据集中的17种蛋白质中,有10种与PBMC数据集重叠。研究团队分别使用sciPENN、Seurat 4和TotalVI方法分析了上述数据(图2)。首先,使用每种方法将PBMC CITE-seq参考数据和MALT scRNA-seq查询数据共同嵌入到一个潜在空间中(图2)。由于PBMC和MALT查询数据之间的巨大差异,即使这三种方法中都采用了内部批量校正策略,sciPENN、TotalVI和Seurat 4仍很难在潜在嵌入空间中完全混合这两个数据集。但sciPENN整合两个数据集的能力最优,其在潜在嵌入中实现了这两个数据集的部分混合。同时,研究团队还检测了三种方法的蛋白表达预测准确性,通过相关性和均方根误差(RMSE)对其进行量化。结果显示,sciPENN在所有蛋白质中实现了最高的蛋白质预测精度。这种高蛋白质预测准确性使sciPENN能够准确地恢复蛋白质表达模式。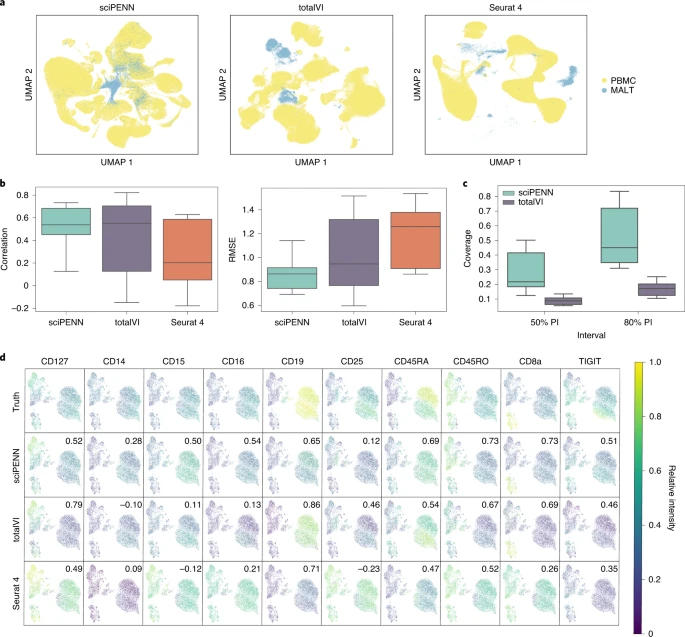
图2. 使用Seurat 4 PBMC数据集作为参考的MALT数据集中的蛋白表达预测。考虑到查询、参考数据集之间更加均衡的平衡,研究团队使用一个人类血液单核细胞和树突状细胞CITE-seq数据集(单核细胞数据集),为测试集保留了真实表达(图3)。分析显示,sciPENN在嵌入过程中实现了两个数据集的完全混合;TotalVI实现了几乎完全的混合,只有极少的不重叠;Seurat 4未完全混合两个数据集。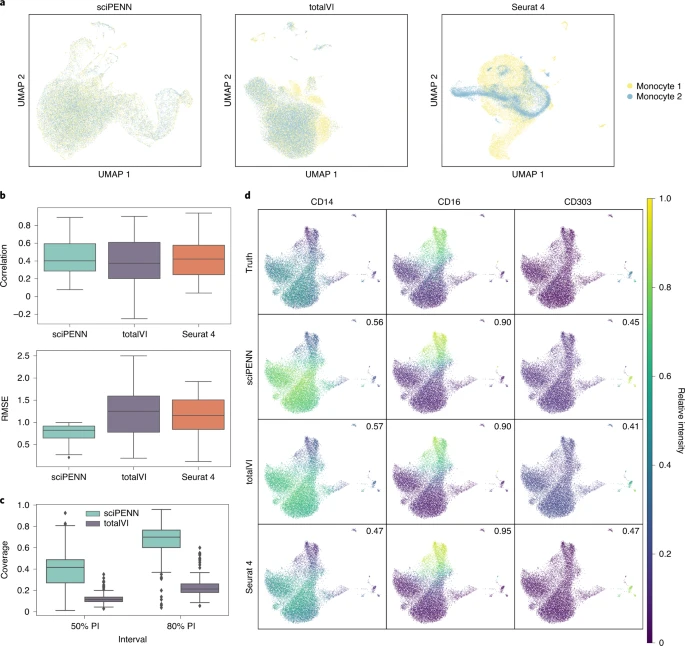
接下来,研究团队随机地将完整的PBMC数据分为训练一半和测试一半,选择了CD8亚型的三种蛋白质标志物(CD45RA,CD44-2和CD38-1)并检测了sciPENN恢复标记蛋白趋势的能力(图4)。CD45RA是CD8幼稚型的明显标记,CD44-2是CD8 TEM3和CD8 TCM2的明显标记,CD38-1是CD8 TCM2的明显标记。结果显示,sciPENN的蛋白质预测准确地恢复了这些趋势,研究人员可仅使用sciPENN预测来检测蛋白质的高表达细胞亚型。TotalVI和Seurat 4的表现比sciPENN略差,Seurat 4低估了CD8 TEM3中CD44-2的表达,TotalVI低估了CD8 NAIVE 2中CD38-1的表达。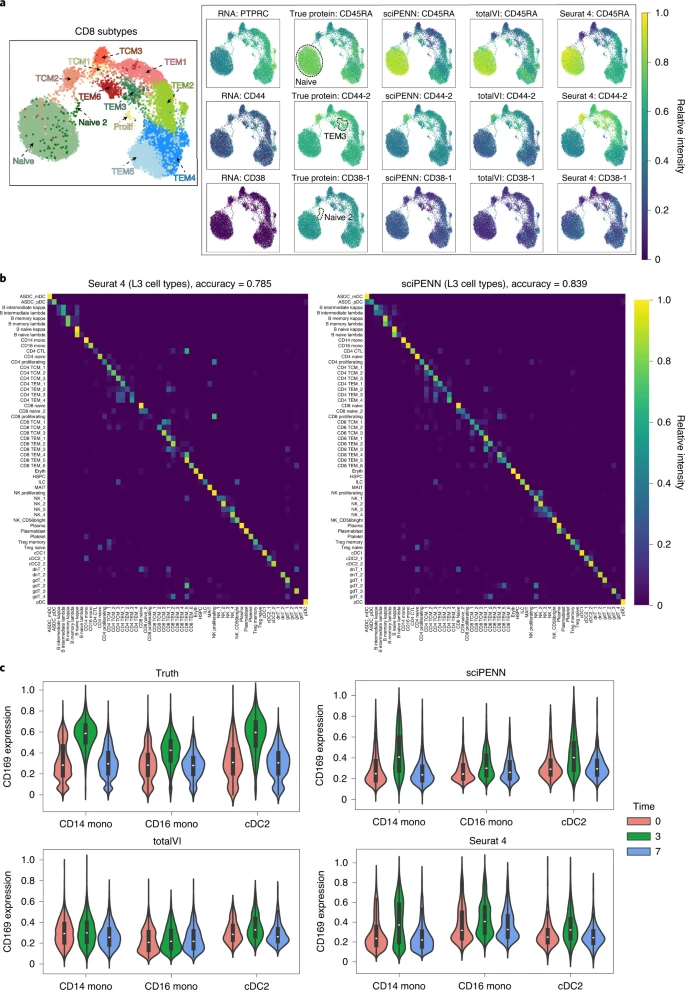
图4. PBMC数据集中的蛋白表达预测和细胞类型标记转移。最后,研究团队检测了sciPENN在PBMC和H1N1 RNA-seq数据中预测蛋白表达能力,由于TotalVI的损失函数迅速衰减为非数字,因此并未将其纳入比较。研究团队将每个测试数据集中预测的蛋白质分为三类:仅存在于Hanifa、仅存在于Sanger和两者都存在。结果显示,与独特蛋白质相比,sciPENN预测常见蛋白质更准确、预测效果越好。上述结果强调了结合多个CITE-seq数据集对蛋白质表达预测的重要性。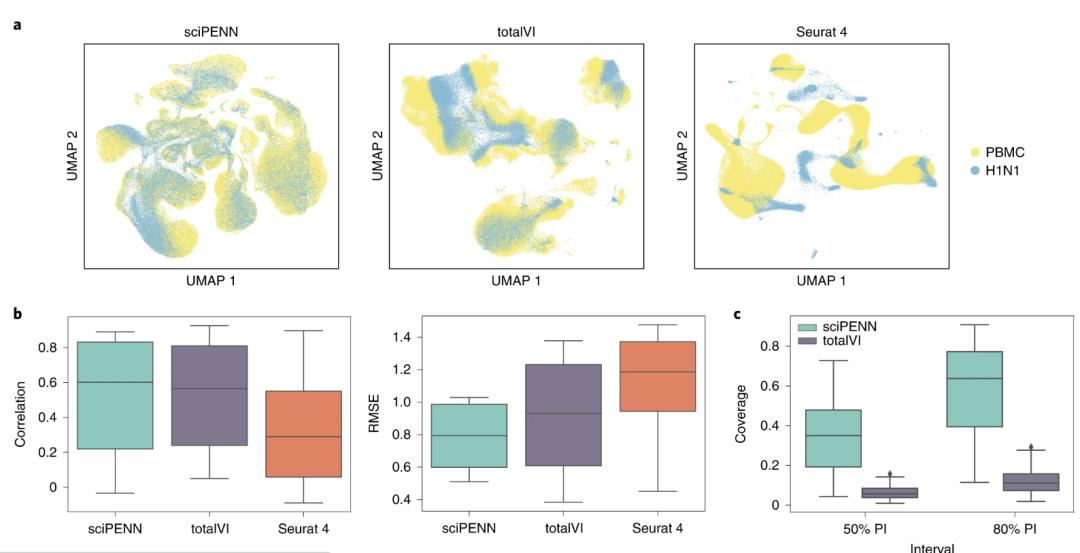
图5. 使用Seurat 4 PBMC数据集作为参考的H1N1数据集中的蛋白质表达预测。综上所述,研究团队开发了sciPENN深度学习模型,可以预测和估算蛋白质表达,集成多个CITE-seq数据集,量化预测和估算不确定性。sciPENN能够从具有部分不重叠蛋白质panel的多个CITE-seq数据集中学习,估算每个组成CITE-seq数据集的缺失蛋白质,甚至在从部分重叠的CITE-seq数据集学习后预测外部scRNA-seq数据集中的蛋白质表达。此外,sciPENN提供了比totalVI和Seurat 4更可靠、准确的结果,同时还具有高度的可扩展性和计算效率,是综合CITE-seq和scRNA-seq数据分析的一个理想工具选择。Lakkis, J., Schroeder, A., Su, K. et al. A multi-use deep learning method for CITE-seq and single-cell RNA-seq data integration with cell surface protein prediction and imputation. Nat Mach Intell (2022). https://doi.org/10.1038/s42256-022-00545-w
·END ·






