Hi,大家好,我是晨曦
上一期推文我们完整的体会了Nature上单细胞分析的基本步骤,可以想到依旧是熟悉的配方,熟悉的味道,首先作者通过对单细胞数据集进行标准流程分析得到了细胞亚群聚类,然后作者本身对巨噬细胞亚群感兴趣,然后就把巨噬细胞亚群进行亚群细分,然后对这些细分的亚群再来进行探索,后续作者还做了针对某一个细胞亚群的细胞轨迹分析,但是这块的部分晨曦还没有整理出来,所以可能需要在下一期再跟大家见面了
但是,本着不能断更且绝不拖更的精神,我们今天依旧是有东西可以进行讨论和学习的,那就是下面这篇今年发表的纯机器学习数据挖掘的Nature(Ps:当时这篇文章出来的时候,很多小伙伴也是私聊晨曦,希望可以出一期解读推文)
那么这期我们就一起来学习一下这篇机器学习数据挖掘的文献吧~
那么,我们开始吧

关于文章的详细内容,笔者在这里就不过多赘述了,因为如果各位小伙伴是挑圈联靠的老粉丝,那么阅读文献想必已经不在话下了,我们把目光重点聚焦在模型构建上,也就是说得到下面这张图的过程:
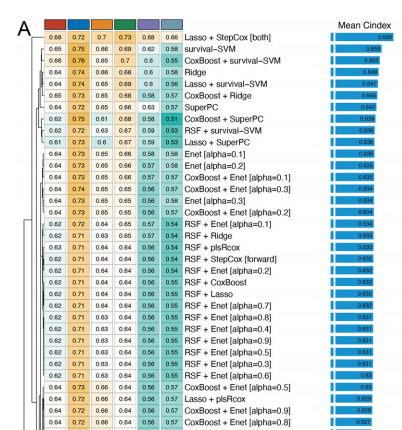
其实简单来说就是,作者通过10种本身具有变量筛选属性的算法构建了一系列算法组合,还记得笔者在以前推文中所叙述的内容吗?
有些模型天生就具有筛选变量的特性
那么很显然,目前已经存在了很多模型,究竟是用模型A筛选变量然后用模型B构建模型比较好,还是用模型B筛选变量然后用模型A构建模型比较好呢?
又或者是干脆用模型C来筛选变量然后用模型A构建模型,然后作者基于这些考量,进行了10种模型不同的两两组合,也就是说会有101种组合,那么很显然,我们既然想要学习这个文献,自然就需要对其中的10种基模型进行学习,因为只有学会了这10种基模型,后续我们只需要构建一个来回交互的循环即可完成上述的分析流程
总结:
如果我们想要复现上述算法或者说是上述的分析流程,我们需要完成下面两个要点:
1.分别构建10种基模型的运算过程
2.设计基模型与基模型之间的来回匹配(也就是说我们需要设计如何让10种基模型之间相互匹配,进而产生101种组合)
那么我们现在就首先来重点了解一下这篇文献所涉及到的10种基模型:
包括:Lasso、Ridge、Enet、StepCox、survivalSVM、CoxBoost、SuperPC、plsRcox、RSF、GBM
1.Lasso
耳熟能详的模型了,基本上我们在进行诊断marker筛选的时候都会想起Lasso,原理部分不是临床工作者深入探索的重点,但是一定程度上了解原理可以让我们更加熟悉算法的运用
Lasso——利用最小二乘法通过引入惩罚系,把不重要的变量系数打压为0来实现筛选变量的目的
A.输入数据要求:一般来说预测变量要求是数值型变量,也就是说如果是字符型最好进行recode
B.关于响应变量用的最多的就是二分类变量,当然笔者是非肿瘤,很少能获得到像作者一样的临床数据,但是上述文章的框架是互通的,也就是说只要我们可以找到10个可以进行二分类变量筛选的预测模型,那么依旧可以使用这个框架,本篇推文也会对此做出探索
C.可以实现此功能的R包包括:glmnet包、mlr3包
#代码解析####1.glmnet包cvfit ####2.mlr3包lrn
2.Ridge
这个模型算的上是Lasso的亲戚了,Lasso是通过添加惩罚项将系数打击为0,而零回归则是通过添加惩罚项将系数不断变小,也就是说不重要的变量的系数会不断变小,但是不会变成0,所以Ridge并不具备筛选变量的能力,但是我们也可以通过对系数的打击而获得比较重要的变量
一般来说在进行机器学习项目的时候,我们都应该把非数值的数据转换为数值数据,因为我们可以这样简单理解,模型本质上其实就是数学公式,之所以有些模型允许我们用字符进行运行,那是因为其模型本身内置的编码,会自动把字符型转换为数值型,但是我们最好自己来转换,因为这样我们可以更加准确的理解结果
#代码解析####1.glmnet包cvfit ####2.mlr3包lrn
3.Enet(弹性网络)
这个模型可以说是Lasso和Ridge的一个集合体,书本上对弹性网络的定义就是兼具Lasso的优点以及Ridge的优点,其实本质上就是通过定义alpha系数的大小来调节惩罚项大小,当然我们也可以直接记忆下面这句话
在 LASSO回归中,我们为 alpha 参数设置一个 '1' 值,并且在 岭回归中,我们将 '0' 值设置为其 alpha 参数。弹性网络在 0 到 1 的范围内搜索最佳 alpha 参数
#代码解析####1.glmnet包cvfit ####2.mlr3包lrn
肯定会有小伙伴问那么应该如何获得最佳的alpha系数,那么这个时候其实解决方法有两个:
1.如果是基于mlr3包的体系那么有自动调优的流程
2.可以直接for循环,把alpha设置为:0.1 —— 0.9
4.StepCox
这一步骤其实就是
两个函数的连用,分别是step函数以及cox函数,简单理解就是通过step函数基于AIC选择一个cox模型,而这个cox模型需要满足AIC最小,那么自然在构建不同的cox模型的时候会选择不同的变量进而获得不同的AIC,所以就获得了变量筛选的能力,那么下面我们来看一下其代码是如何实现的:
#代码解析####1.survival包for (direction in c("both", "backward")) { fit ####2.mlr3包#这个目前在mlr3中并不可以得到实现,所以我们只有采用基本函数来实现这个应用
5.survivalSVM
支持向量机模型算得上是平时基于R完成机器学习中我们比较熟知的模型了,因为在以前晨曦也更新过相关的推文来介绍SVM,其实简单理解就是通过添加更高维度的超平面然后实现数据的划分,但是针对于生存数据,或多或少会有所不同,但是核心是相同的
#代码解析survivalsvm(Surv(OS.time,OS)~., data= est_dd, gamma.mu = 1)##核心函数
6.CoxBoost
单纯的cox模型因为属于半参数统计模型,所以模型的可解释性较强,但是预测性能往往会低于非参数化模型,所以这个时候我们想要对这个模型进行优化,那么可以参考机器学习中集成学习模型的思路,而集成学习思路包含:boosting、bagging以及不常用的Stacking,那么这里显然就是COX + boosting
#代码解析#github上有现成的R包可以完成这项操作library(devtools)install_github("binderh/CoxBoost")RIF mix.list=c(0.001, 0.01, 0.05, 0.1, 0.25, 0.35, 0.5, 0.7, 0.9, 0.99), stratum=group,stratnotinfocus=0,penalty=sum(obs.status)*(1/0.02-1), criterion="hscore",unpen.index=NULL) #当然作者针对这里使用的是下面这套代码fit stepno=cv.res$optimal.step,penalty=pen$penalty)
7.SuperPC
这个算法的中文名称叫做监督主成分分析,其实这个算法笔者也是通过这篇文献第一次知道,对于其性能在使用的时候感觉并不是十分的惊艳,但是主成分分析我们都知道,所以其实在笔者看来应该是把主成分分析与有监督学习进行了结合最后得到了这个算法,相关的算法也在github上有大佬的复现:jedazard/superpc: Supervised Principal Components (github.com)
题外话:笔者把R语言的境界概括为以下三个阶段:
1.入门(这个阶段停留在软件不会用、代码看不懂)
2.中阶(这个阶段为代码能看懂,能够改代码,能够做到只要网络上有教程就可以去用)
3.高阶(自己编写算法和代码)
目前笔者仍然停留在第二个层次,但是通过这一点笔者想要告诉小伙伴的是只要我们有一个普遍的需求,我们完全可以通过网络找到答案而并不是完全依靠自己从零开始,借鉴网络上优秀的教程进行学习可以提高我们进步的速度QAQ
#代码解析cv.fit n.fold = 10,n.components=3,min.features=5,max.features=nrow(data$x),compute.fullcv= TRUE,compute.preval=TRUE)
需要注意的是,本篇文献的作者基本上都是使用交叉验证来构建模型或者来评估模型等,所以说最后构建好的最佳模型组合也被作者命名为:leave-one-out cross-validation (LOOCV) framework
8.plsRcox
plsRcox 实现了偏最小二乘回归和各种内核技术,其用途是主要用于在高维设置中拟合 Cox 模型,那么如果作为临床科研工作者,我们只需要简单理解,高维数据中如果带有生存预后信息,那么可以通过plsRcox这个算法实现在高维数据中拟合cox模型
9.RSF
这个其实就是我们常说的随机生存森林,那么其实简单理解就是可以运用在生存数据中的随机森林模型,而随机森林模型本身就是集成学习模型的一个代表,而且其性能其实也是非常的出众,又因为其对输入数据要求比较低,所以也是很受人欢迎
通过“randomForestSRC”包中的rfsrc函数构建随机生存森林模型
#代码解析fit ntree = 1000,nodesize = rf_nodesize,##该值建议多调整splitrule = 'logrank',importance = T,proximity = T,forest = T,seed = seed)
10.GBM
GBM其实在笔者看来运用的算法其实和XGboost是相似的,都是串联的生成一系列弱学习器,然后每个弱学习器的目标是拟合先前累加模型的损失函数,也就是学习前面的错误
#代码解析fit n.trees = 10000,interaction.depth = 3,n.minobsinnode = 10,shrinkage = 0.001,cv.folds = 10,n.cores = 6)
至此,10种基模型就给大家简单的过了一遍,看到这里的小伙伴可能会发现,我们只需要整理好输入数据其实就可以调用算法,而这篇文章在于选择最合适的组合来获得最佳的biomarker的同时构建预测模型
那么这篇文章其实是包含了生存数据的,但是很多小伙伴其实是非肿瘤,那么也可以参考上面的流程构建自己的诊断模型,比如说纳入Lasso、对数几率比回归、SVM、随机森林、XGboost、岭回归、弹性网络,然后根据上面的流程走一遍也是可以完成分析的
那么问题其实在于我们应该如何批量进行操作,鉴于笔者刚刚接触这篇文章,目前能想到一定可以结局的方法如下:
1.101种可能其实就是相当于跑101便流程,肯定是可以通过批量循环进行操作,但是如果单纯通过自己手动运算,工作量其实也不是十分巨大,首先列出一个所有组合的Excel表格,然后一步步去运行是肯定可以完成的
2.编写循环函数的关键在于对不同的组合运行不同的函数,所以这里笔者暂时能想到的这套流程的关键函数可能是——if、for以及do.call函数
初步设想的流程可能是,首先我们依旧是列出所有的可能,然后分别编写每个基模型的函数,然后通过do.call函数进行不同分支选择的执行情况,当然貌似这个代码编写还是比较困难的,笔者会钻研一段时间!
这个分析流程其实给了我们很多的启示,通过这样的组合可以让自己的SCI增加很多的工作量,而且如果一次次操作代码重复运行其实也是机械性的工作了,性价比还是挺高的~
当然作者本人也是上传了每个基模型的代码,感兴趣的小伙伴可以上作者的github上进行学习,最后在使用这项技能的时候也别忘了引用作者文献哦(感谢作者开源的精神QAQ)
Machine learning-based integrative analysis develops an immune-derived lncRNA signature for improving clinical outcomes in colorectal cancer
本期推文到这里就结束啦
我是晨曦,我们下期再见~









