
化学作为在原子、分子水平上研究物质的组成、结构、性质、转化及其应用的基础自然科学,其源自生活和生产实践,并随着人类社会的进步而不断发展。其中,化学知识是多方面的,在分析化学结构的特定性质时,有必要考虑各种尺度,比如特定的片段、键以及原子,并且这些局部属性能够为结构的全局属性和功能提供重要见解。原子尺度上的局部性质(如原子电荷和杂化)和分子尺度上的全局性质(如偶极矩、基态和激发态能)已成为化学思维和描述基本概念和设计应用的实用词汇的核心。随后,这些性质成为实验和理论研究的主要目标。所有这些性质都可以从计算求解薛定谔方程的第一性原理电子结构计算中推断出来。然而,在实践中,很少提出精确的解决方案。因此,之前的研究已经开发出一系列具有不同保真度的方法,从非常精确的波函数方法(如耦合簇技术)到实用和广泛使用的密度泛函理论(DFT),再到精度较低的半经验方法。即使经过几十年的发展,传统电子结构理论的适用性仍然受到基础数值方法的基本尺度的限制。如今,机器学习(ML)已经成为复杂化学过程和材料建模的首选策略,其提供了一个在参考数据集上训练的替代模型,可用于构建分子结构与其化学性质之间的相关性,可以为大规模系统提供快速解决方案。化学ML的进步清楚地表明,原子和分子特性可以被机器“学习”,从而有可能克服上述传统的限制和数字障碍。
在此,美国洛斯阿拉莫斯国家实验室Sergei Tretiak教授(通讯作者)全面总结了基于ML评估化学性质的发展,例如部分原子电荷、偶极矩、自旋、电子密度、化学键以及获得简化的量子力学描述。同时,作者概述了几种现代神经网络架构,以及它们的预测能力,通用性和可转移性,并说明了它们对各种化学性质的适用性。此外,作者还强调了学习的分子表示类似于量子力学类似物,证明了模型捕获基础物理学的能力,以及讨论了ML模型如何描述非局部量子效应。最后,作者编制了一份可用的ML工具箱列表,总结了未解决的挑战和展望了未来的发展。总结的趋势表明,本领域的发展正在向由ML增强的基于物理的模型发展,其伴随着新方法的开发以及用户友好的ML化学框架正在快速增长。
相关研究成果以“Extending machine learning beyond interatomic potentials for predicting molecular properties”为题发表在Nature Reviews Chemistry上。

1.本文概述了一组快速出现的简化量子化学方法,例如密度功能紧密结合(DFTB),并指出了ML如何直接改善电子结构计算;
2.作者编制了一份可用的ML工具箱列表,总结了未解决的挑战和展望了未来的发展,证明了本领域的发展正在向由ML增强的基于物理的模型发展。
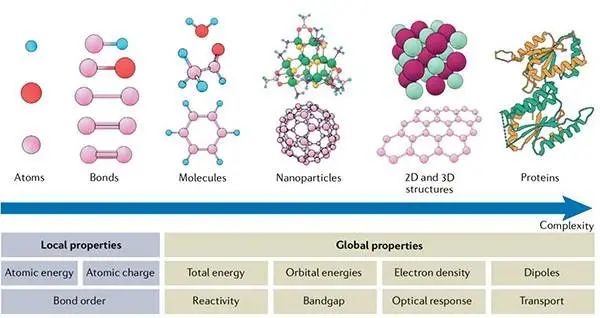
图1从化学家的角度看物质的原子尺度©2022 Springer Nature
局部性质与原子、键或碎片等基本的结构元素有关,而全局性质则归属于整个系统。二维(2D)和三维(3D)结构通常被视为具有周期性,因此蛋白质作为一个整体非周期的例子,其复杂性规模在不断增加。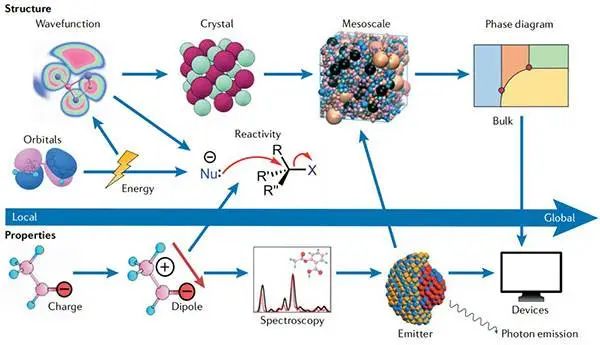
图2从局部和整体角度看化学结构与性质的关系©2022 Springer Nature
相互关系性是直观的,但不是严格限定的。例如,电荷和偶极子是由量子力学波函数来定义,这些系统的动力学反映在可测量的光谱中,并支持理想的特性,如光子的发射。
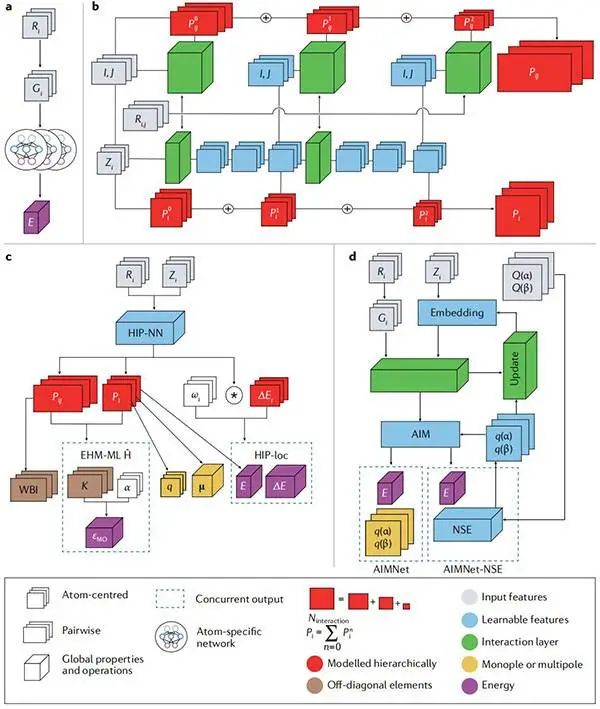
图3用于学习局部和全局属性的神经网络的现代体系架构©2022 Springer Nature
(a)高维神经网络(HDNN)的分子能量(E)预测;
(b)层次结构相互作用的粒子神经网络(HIP-NN)体系结构;
(c)HIP-NN变体用于学习不同的原子和分子性质;
(d)分子中原子网络(AIMNet)体系结构的变体;
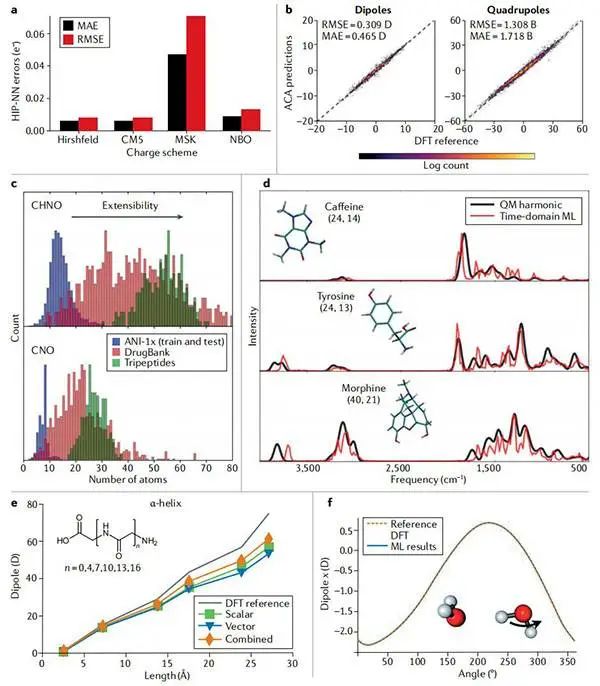
图4原子电荷、振动谱、偶极子和四极子的机器学习预测©2022 Springer Nature
(a)使用测试集(ANI-1x)训练以重现各种电荷分配方案的部分原子电荷时,分层相互作用粒子神经网络(HIP-NN)电荷预测的平均绝对误差(MAE)和均方根误差(RMSE);
(b)与密度泛函理论(DFT)参考文献相比,当仅在ANI-1x偶极子上进行训练时,HIP-NN平价电荷分配(ACA)模型在三肽集上的性能;
(c)ACA模型的训练和扩展性集中按大小分布的分子。顶部面板计算每个分子的原子总数(C,H,N,O),而底部面板计算每个分子的非氢原子(C,N,O)的数量;
(d)选定生物活性分子的红外光谱的ACA模拟;
(e)机器学习(ML)预测了不同链长的α-螺旋构象中聚甘氨酸的偶极矩,并与DFT参考文献进行了比较;
(f)在二聚体中,水分子中O-H键旋转时向x轴投射的偶极矩的动力学;
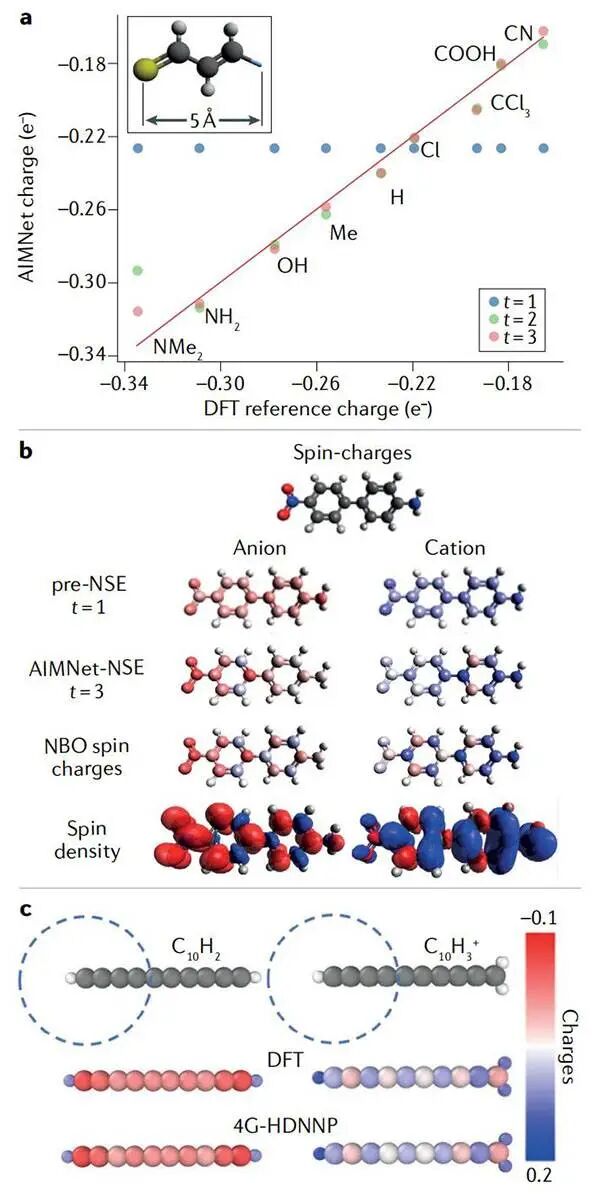
图5自旋极化电荷和总电子密度的机器学习预测©2022 Springer Nature
(a)一系列取代的硫代醛中硫原子上的原子电荷,正如在 ANI-1x 数据集上训练的分子中原子网络(AIMNet)所预测的那样,该网络由氟、硫和氯原子的分子增强;
(b)在AIMNet神经自旋平衡(AIMNet-NSE)中的迭代更新使同时学习α和β的电子密度成为可能;
(c)第四代(4G)高维神经网络(HDNN)电位在模拟非局部电荷传输中的性能。
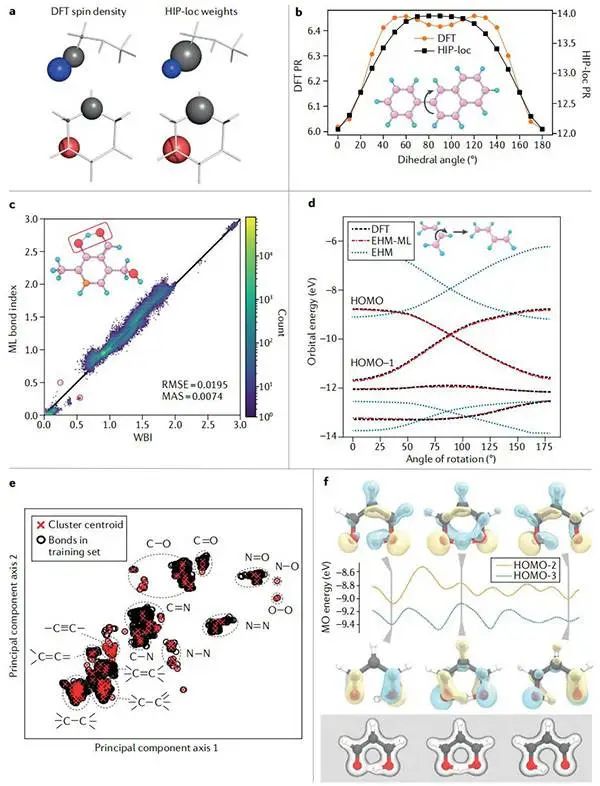
图6自旋密度、键序和有效哈密顿模型的机器学习预测©2022 Springer Nature
(a)比较所选分子的密度泛函理论(DFT)自旋密度和可视化分层相互作用粒子定位(HIP-loc)定位权重;
(b)扫描多环芳烃分子中C-C二面角时参与率(PR)的变化;
(c)在ANI-1x集的一部分上训练并应用于Drugbank集的HIP-NN模型的性能;
(d)1,3-丁二烯顺反异构化过程中的前沿分子轨道(MOs)交换;
(e)在训练集中具有不同键拓扑的聚类,用于拟合密度泛函紧密结合(DFTB)框架中的键特异性排斥势;
(f)质子转移在丙二醛中作为分子动力学模拟的一系列快照。
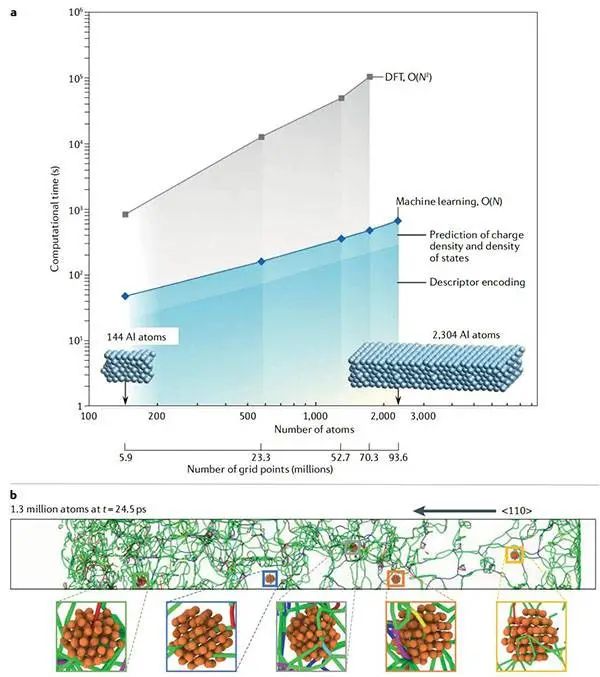
图7通过机器学习实现的大规模分子模拟©2022 Springer Nature
(a)密度泛函理论(DFT)与机器学习(ML)的比例比较;
(b)在应用元素铝(ANI-Al)电位后,在24.5ps的冲击下,使用ANI模拟了铝体相的位错结构。
综上所述,受益于高质量的数据,数据驱动的主动学习 技术为原子间势数据集的最佳收集提供了有前途的策略,而不会失去其可转移性。目前,许多架构要么在一个目标属性上训练,要么调用单独的网络,每个网络负责自己的标签。总而言之,计算成本仅是分子力学的十倍。一个趋势是明确的:ML方法正在成为计算科学家工作台上不可或缺的工具。ML正在成为经过时间验证代码的重要组成部分。例如,最新版本的ORCA 5.0引入了ML优化的DFT集成网格。此外,基准测试明确展示了热化学、反应势垒、非共价相互作用和振动频率评估方面的改进,体现了纯电子结构计算如何从数据科学中受益。
文献链接:“Extending machine learning beyond interatomic potentials for predicting molecular properties”(Nature Reviews Chemistry,2022,10.1038/s41570-022-00416-3)
本文由CYM编译供稿。
如果您希望开展计算工作,苦于没有合适的计算人才,也可以联系我们。6年以来,我们与入驻材料人平台的300余位计算顾问一起,共同服务了超过5000位客户,涵盖了包括VASP、Lammps等在内的多款软件以及第一性原理、分子动力学、有限元等领域。










