本文介绍吉林大学李向涛教授课题组发表在Advanced Science的研究成果,题为“Distribution-Agnostic Deep Learning Enables Accurate Single-Cell Data Recovery and Transcriptional Regulation Interpretation”。单细胞转录组测序(scRNA-seq)是一种在单细胞水平上研究基因表达的可靠方法,但是准确的量化转录信息通常受到有限的mRNA捕获的阻碍,从而导致许多缺失的表达值。现有的插补方法依赖于严格的数据假设,限制其更广泛的应用,从而导致有偏的信号恢复。为了应对这一挑战,作者提出了一个分布无关的深度学习模型,可准确恢复缺失的基因表达。该模型基于最优传输理论,通过正则化细胞嵌入空间来应对单细胞转录组数据的复杂分布。此外,还提出了表达一致性模块引入bulk RNA-seq数据指导缺失基因恢复。
所提出的方法(图1)不依靠假设的任一概率分布,而是根据最优传输理论考虑未知的真实数据分布与重建数据分布之间的最小化差异。同时,该模型有效地采用了细胞嵌入空间正则化来捕获细胞之间的几何和拓扑关系。这种策略方法确保了在生物学上相关性的提取,同时最大程度地减少了无关紧要的噪声。此外,解码器对此过程中捕获的细胞间相关性进行了高效利用,以执行准确的缺失数据恢复。同时,Bis结合了转录表达一致性模块,利用匹配的bulk RNA-Seq和scRNA-Seq数据之间基因平均表达的一致性监督缺失信号恢复。在多个真实和模拟的数据集中,Bis的插补性能优于现有方法,并显著提高了下游分析性能,包括批次效应校正,聚类分析,差异表达分析和轨迹推断。此外,Bis准确重建基因表达模式的能力协助在头颈肿瘤匹配的外周血液数据集中识别出了稀有细胞类型,揭示了对复杂的头部和颈部鳞状细胞癌(HNSCC)微环境中细胞因子诱导的记忆样自然杀伤细胞的发育成熟机制。
作者在8种不同丢失率(26%到93%)的仿真数据集和4个不同规模的真实scRNA-seq数据集上进行了多个实验,与当前先进的多个插补算法进行了性能比较,包括ALRA、DCA、DeepImpute、MAGIC、SAVER、scImpute、SCRABBLE、scScope和scVI。为了准确全面的评估算法插补的表现,作者采用了细胞间和基因间的皮尔逊系数以及其均值和方差这几个指标。结果表明Bis仿真和真实数据集上都优于对比算法,显著提高了数据质量。与基于特定概率分布假设的算法相比,Bis在指标提升上也表现出一定的优势。这表明Bis在scRNA-seq数据插补方面具有更高的准确性和鲁棒性。
Bis提升了细胞类型识别和scRNA-seq数据可视化
聚类是分析scRNA-seq数据的关键步骤,其揭示了组织系统的复杂性(例如,识别细胞类型和研究细胞异质性)。作者在多个不同规模的scRNA-seq数据集上进行了聚类分析,以间接验证插补算法的有效性。随后作者统计了ARI、NMI和Jaccard三个常用的聚类指标,结果如图2A所示,Bis在大多数数据集上始终优于对比方法。此外,与其他方法相比,Bis在这四个数据集上始终表现出卓越的性能。
另外,图2B展示了在肾脏数据集上经不同插补算法处理后的2维UMAP结果。经Bis处理后的数据在可视化质量方面表现出非凡的性能,尤其是在细胞种群紧密相结合且难以区分的情况下。比如在肾脏数据集中,Bis的结果成功地识别了远端收集导管主细胞种群的两个HSD11B2和CLDN4高表达的亚群(图2B),但是,其他算法倾向于将这两个细胞亚型与其他细胞类型误分类。
图2 Bis提升了细胞类型识别和scRNA-seq数据可视化
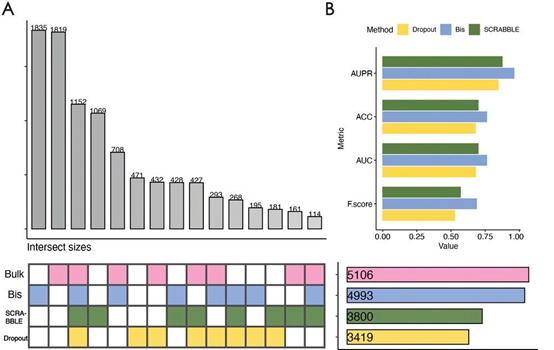
差异表达分析旨在确定表达水平通过特定变量(例如细胞类型或状态)显著改变的基因。这些差异表达基因的鉴定提供了对表型变异的基本分子机制的宝贵见解。但是,差异表达分析方法的结果易受scRNA-seq数据中表达丢失基因的影响。与bulk RNA-seq数据相比,原始的scRNA-seq数据表现出基因零表达的比例较高,并且与bulk数据共享了较少数量的差异表达基因(图3A)。但是,在对原始的scRNA-seq数据执行插补操作后,差异基因的数量显著增加,并且接近bulk数据中鉴定的差异基因数量。同时,作者采用了多个评价指标,包括Precision-Recall曲线(AUPR),准确性(ACC),F-SCORE(也被认为是F1得分或F-measure)和接收者操作特征曲线下的面积(AUC),以评估差异表达基因检测性能,Bis在所有指标中都取得了最高的分数。随后,作者对Bis恢复出的特异性差异基因进行了生物学相关性探索,发现这些差异基因在与胚胎细胞发育、干性和分化相关的通路中显著富集。
Bis揭示细胞因子诱导的记忆样自然杀伤细胞在循环免疫微环境中的发育特征
在对Bis与其他对比方法进行了全面评估后,作者探索 了Bis的重建能力是否可以增加生物发现的可能。作者将Bis应用于包括36030个新切除的头颈部鳞状细胞癌(HNSCC)肿瘤细胞的外周血数据集。该队列包括17例未经治疗的患者(6例人乳头瘤病毒(HPV)+和11例人乳头瘤病毒)。在该数据集的原始研究中[2],自然杀伤细胞被识别为CD56dimCD16+CD57- 表型且被分为两个细胞亚型。经过插补重建后的数据,除了以上两个细胞亚型被识别,还鉴定出一个新的细胞亚型。通过时序分析,作者发现新识别的细胞亚型代表了细胞因子诱导的记忆样自然杀伤细胞从初始到成熟状态发展中的一个中间状态(图4)。同时,该细胞亚型与已有关于自然杀伤细胞的研究[3]定义的hNK_Bm4亚型也是一致的。
图4. 揭示细胞因子诱导的记忆样自然杀伤细胞在循环免疫微环境中的发育特征
在本文中,作者利用最优传输理论最小化未知真实数据分布与重构数据分布之间的差异提出了一个分布无关的scRNA-seq数据深度插补模型。同时,该模型可以利用bulk RNA-seq数据的外部先验来完善重建过程。模拟和真实数据集的全面研究表明,与原始scRNA-seq数据相比,Bis生成的恢复数据表现出增强的细胞类型识别和差异基因检测,因此在下游分析中得出了卓越的结果。在分析肿瘤匹配的外周血数据集时,该模型提供了更详细的细胞因子诱导的记忆样自然杀伤细胞发育状态的描写。
Y. Su, Z. Yu, Y. Yang, K.-C. Wong, X. Li, Distribution-Agnostic Deep Learning Enables Accurate Single-Cell Data Recovery and Transcriptional Regulation Interpretation. Adv. Sci. 2024, 2307280. https://doi.org/10.1002/advs.202307280
Kürten, Cornelius HL, et al. "Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing." Nature communications 12.1 (2021): 7338.
Crinier A, Dumas P Y, Escalière B, et al. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia[J]. Cellular & molecular immunology, 2021, 18(5): 1290-1304.
https://github.com/XuYuanchi/Bis









