
作者 | Andre Ye
译者 | 孟翔杰
来源 | DeepHub IMBA
许多人没有想到,病毒就像地球上为生存而挣扎的其他生物一样,它们会进化或变异。
只要看一看人类病毒来源的蝙蝠携带的病毒RNA序列片段即可。
AAAATCAAAGCTTGTGTTGAAGAAGTTACAACAACTCTGGAAGAAACTAAGTT
…以及人类COVID-19病毒的RNA序列的摘录…
AAAATTAAGGCTTGCATTGATGAGGTTACCACAACACTGGAAGAAACTAAGTT
…显然,冠状病毒已改变其内部结构以适应其新宿主(更精确地说,冠状病毒内部结构的约20%发生了突变),但仍保持了足够的活力,以至于它仍然可以在起源物种体内存活。
实际上,研究表明COVID-19通过反复变异来提高它们的存活率。在抗击冠状病毒的斗争中,我们不仅需要找到消灭病毒的方法,还需要找到病毒如何突变以及如何遏制这些突变的方法。
在本文中,我将……
提供RNA序列的简单解释
使用K-Means创建基因组信息集群
使用PCA可视化集群
…并对我们执行的每个程序进行分析来获取经验。
与“解码”相比,基因组测序通常是分析从样品中提取的脱氧核糖核酸(DNA)的过程。在每个正常细胞内有23对染色体,这些染色体容纳着DNA。DNA的卷曲双螺旋结构使其可以展开为梯形。该梯形由成对的化学字母(称为碱基)制成。DNA中只有四种:腺嘌呤,胸腺嘧啶,鸟嘌呤和胞嘧啶。腺嘌呤仅与胸腺嘧啶结合,而鸟嘌呤仅与胞嘧啶结合。这些碱基分别用A,T,G和C表示。
这些碱基构成了指示生物体如何构建蛋白质的各种代码-实际上是控制病毒行为的DNA。
DNA转换为RNA再转换为蛋白质的过程
使用包括测序仪器和专用标签等专用设备,可以揭示特定片段的DNA序列。从中获得的信息将用于进一步的分析和比较,以使研究人员能够识别基因的变化过程,并将基因与疾病和表型以及潜在的药物靶细胞关联起来。
基因组序列是一串长长的“ A”,“ T”,“ G”和“ C”,代表生物体对环境的反应方式。通过改变DNA可以造成生物的突变。查看基因组序列是分析冠状病毒突变的有力方法。
了解数据
在Kaggle上找到的数据如下所示:
每行代表蝙蝠病毒的一种突变。首先,只需花一分钟就可以欣赏大自然的不可思议性-在数周之内,冠状病毒已经产生了262种自身突变,以提高自身存活率。
一些重要的列:
query acc.ver 代表原始病毒标识符。
subject acc.ver是病毒突变的标识符。
% identity 代表当前序列与原始病毒相同的百分比。
alignment length 表示序列中有多少个相同的标识符。
mismatches 代表突变和原始变异的数量。
bit score 表示衡量对齐程度的度量;分数越高,对齐越好。
每列的一些统计度量(可以在Python中使用data.describe()方便地调用它):
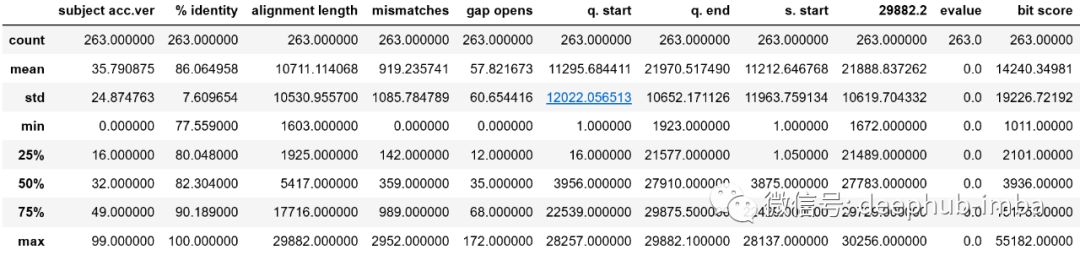
查看% identity列,很有趣的是,该值最小约为77.6%。 % identity的标准差为7%。这个数值相当大,意味着存在广泛可能的突变。 bit score比较大的标准差支持这一观点-标准差大于平均值!
可视化数据的一种好方法是通过关联热图。每个单元代表一个特征与另一个特征之间的关联程度。
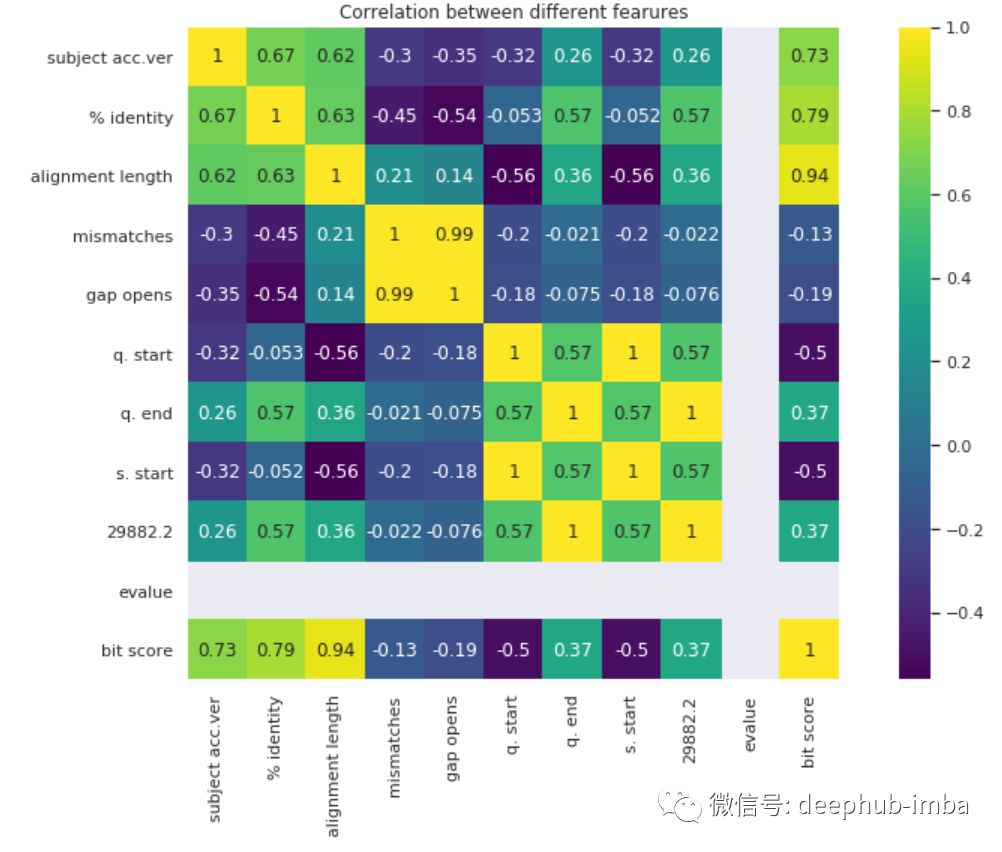
图中可以看到许多数据彼此高度相关。这是有道理的,因为大多数突变都是彼此不同的。需要注意的一件事是alignment length与bit score高度相关。
 使用K-Means创建突变聚类
使用K-Means创建突变聚类
K-Means是用于聚类的算法,它是机器学习中在特征空间中查找数据点并结合成组的一种方法。我们的K-Means的目标是找到突变簇,由此我们可以得出有关的突变性质以及如何解决突变的见解。
但是,我们仍然需要选择簇数k。尽管这就像在二维中绘制点一样简单,但在更高的维度中则无法实现(如果我们想保留最多的信息)。像肘法(elbow method)这样选择k的方法是主观且不准确的,因此,我们将使用轮廓法(silhouette method)。
轮廓法是针对k个聚类中心给出的聚类结果对数据适应程度的评分。Python中的sklearn库使实现K-Means和轮廓法变得非常简单。
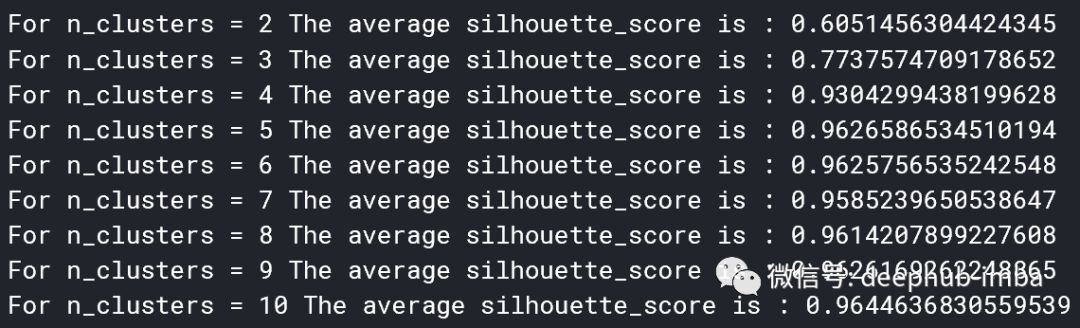
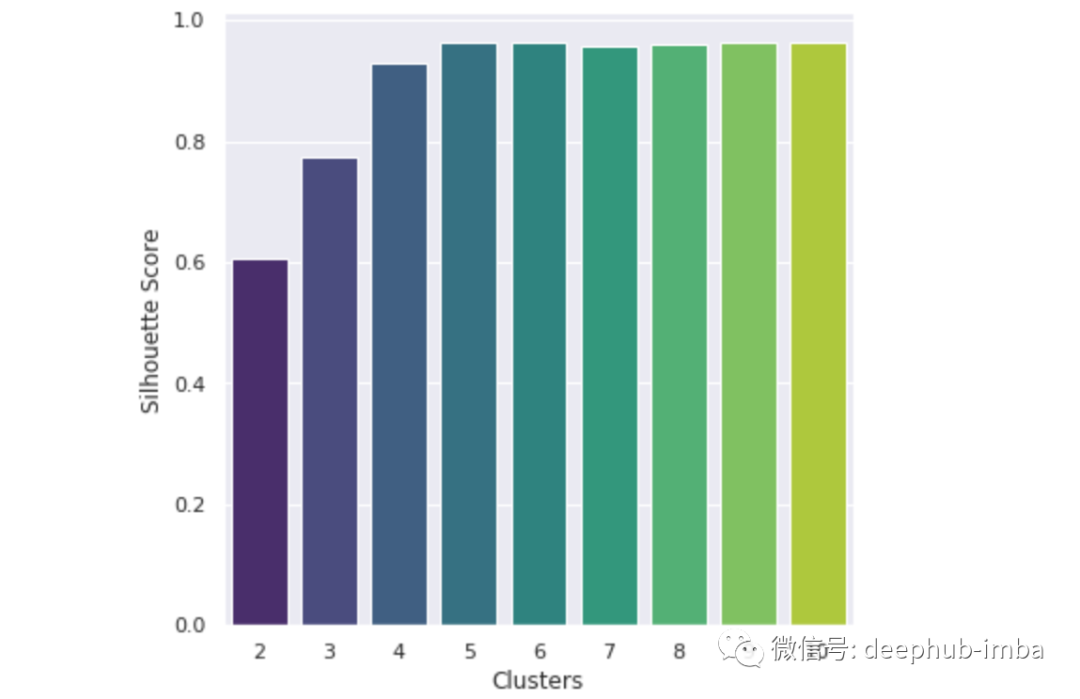
似乎5个聚类中心是最合适的。现在,我们可以确定聚类中心。这些聚类中心是每个类别围绕的点,代表(在这种情况下)5种主要突变类型的数值评估。
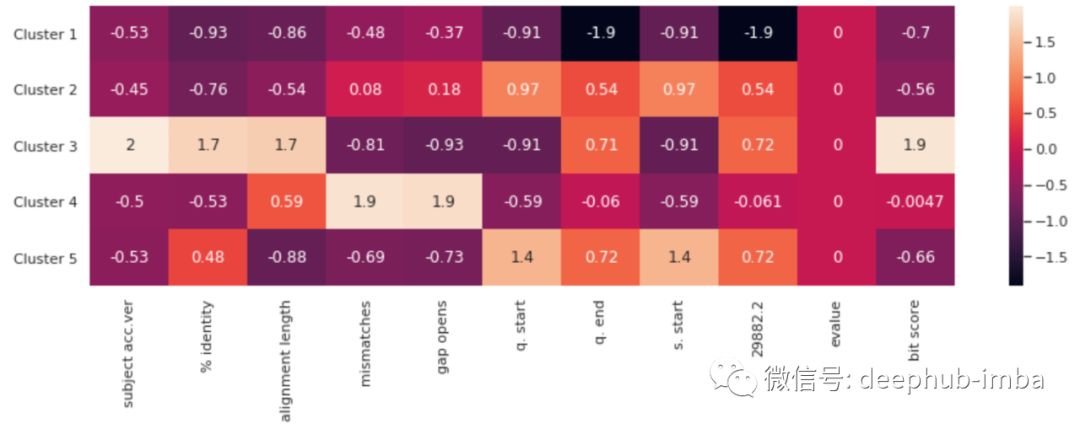
注意:已对数据进行了标准化,以使它们全部缩放为相同的比例。否则,每列将不可比较。
此热图按列表示每个簇的属性。因为这些点是按比例缩放的,所以图中标注的数值在数量上没有任何意义。但是,可以比较每列中的标注值。您可以从视觉上了解每个突变簇的相对属性。如果科学家要开发疫苗,它应针对这些主要的病毒突变簇。
在下一节中,我们将使用PCA可视化数据。
PCA数据可视化
PCA(主成分分析法)是一种降维方法。它选择多维空间中的正交矢量来表示轴,从而保留了最多的信息(方差)。
流行的Python库sklearn可以用两行代码实现PCA。首先,我们可以检查数据的方差比。这是从原始数据集中保留的统计信息的百分比。在这种情况下,数据的方差比是0.9838548580740327,这已经很高了!我们可以放心,无论我们从PCA进行的任何分析都不会使数据失真。
每个新功能(主要组件)都是其他几列的线性组合。我们可以通过热图可视化其中一列对于相对的两个其他组件之一的重要性。
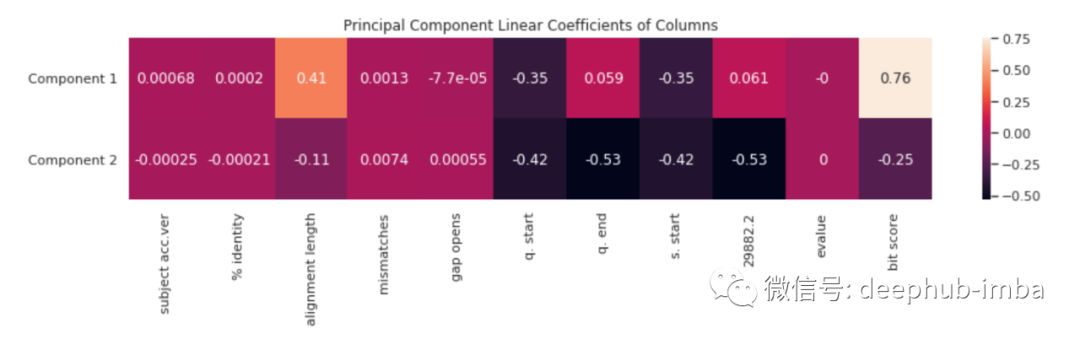
主要需要了解成分一具有较高分数的含义-在这种情况下,其特征具有更长的比对长度(更接近原始病毒),而成分2的主要特征具有更短的比对长度 (距离原始值更远)。这也反映在bit score的较大差异上。
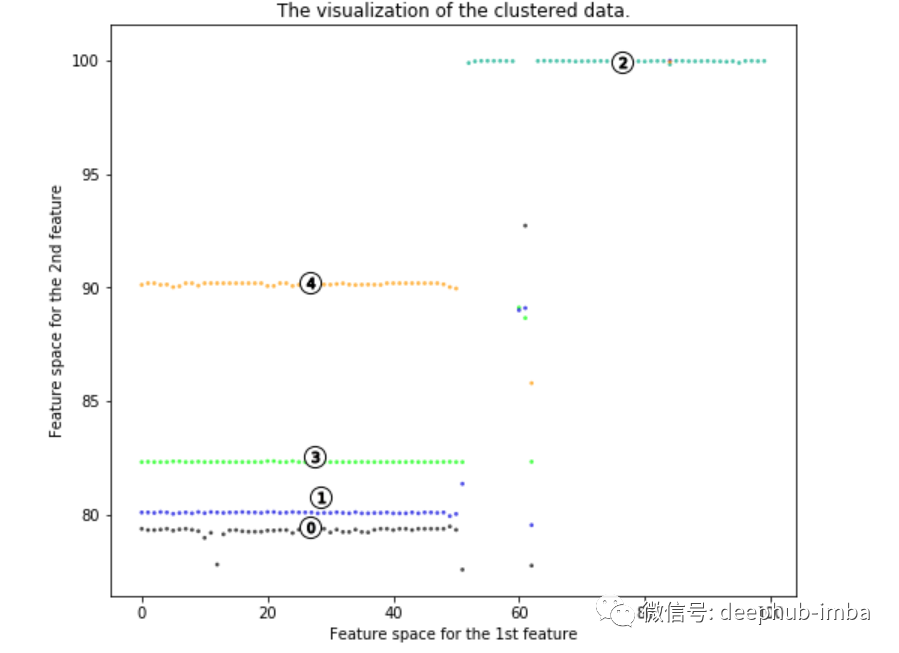
显然,病毒突变有5种主要方式。我们可以从中获得许多信息。
其中四个突变位于成分一的左侧,一个突变位于右侧。成分一的特征是高alignment length。这意味着成分一的值越高则对齐长度越长(更接近原始病毒)。因此,成分一的值较低时在遗传上距离原始病毒较远。因此大多数病毒突变与原始病毒差异很大。因此,试图制造疫苗的科学家应该意识到该病毒会产生大量与原始病毒差异很大的突变。
结论
使用K-Means和PCA,能够识别冠状病毒中的五个主要突变簇。研发冠状病毒疫苗的科学家可以利用聚类中心的信息获得有关每个聚类特征的知识。我们能够使用主成分分析在二维空间上可视化簇,并发现冠状病毒具有很高的突变率。这可能是它如此致命的原因。








