基因组结构元件的可视化有多种方式,比如IGV等基因组浏览器中以track为单位的展示形式,亦或以circos为代表的圈图形式,比如在细胞器基因组组装中,基因元件常用圈图形式展示,示例如下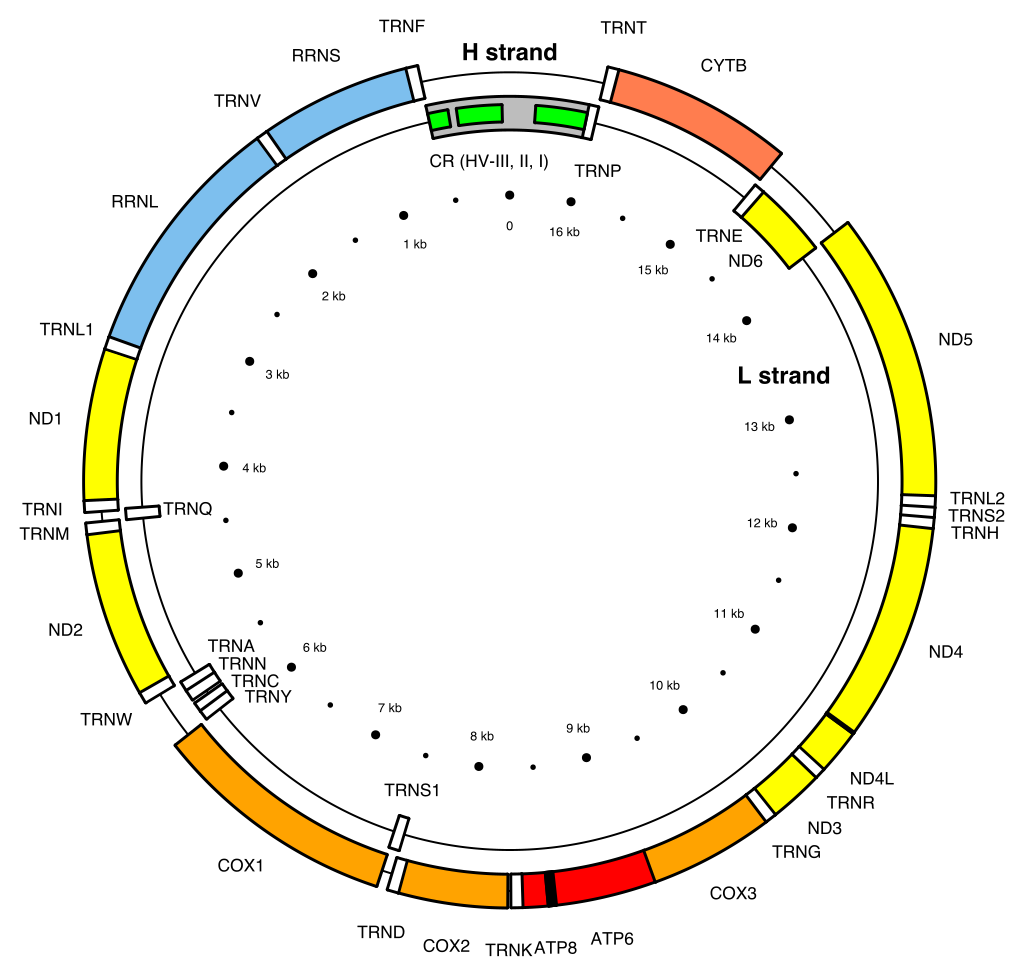
在biopython中,通过BiolGraphics子模块可以对基因组结构进行可视化,支持线性和圈图两种可视化方式。其中,基因组结构信息存储在genebank格式的文件中,首先通过Bio.SeqIO读取结构信息,然后通过Bio.Graphics模块进行可视化。
以下列数据为例,先来看下可视化的用法
>https://www.ncbi.nlm.nih.gov/nuccore/NC_005816
首先是读取gb文件,代码如下
>>> from reportlab.lib import colors
>>> from reportlab.lib.units import cm
>>> from Bio.Graphics import GenomeDiagram
>>> from Bio import SeqIO
>>> record = SeqIO.read("sequence.gb", "genbank")
接下来提取gb文件中的feature信息,构建用于绘图的数据结构,代码如下>>> gd_diagram = GenomeDiagram.Diagram("Yersinia pestis biovar Microtus plasmid pPCP1")
>>> gd_track_for_features = gd_diagram.new_track(1, name="Annotated Features")
>>> gd_feature_set = gd_track_for_features.new_set()
>>> for feature in record.features:
... if feature.type != "gene":
... continue
... if len(gd_feature_set) % 2 == 0:
... color = colors.blue
...
else:
... color = colors.lightblue
... gd_feature_set.add_feature(feature, color=color, label=True)
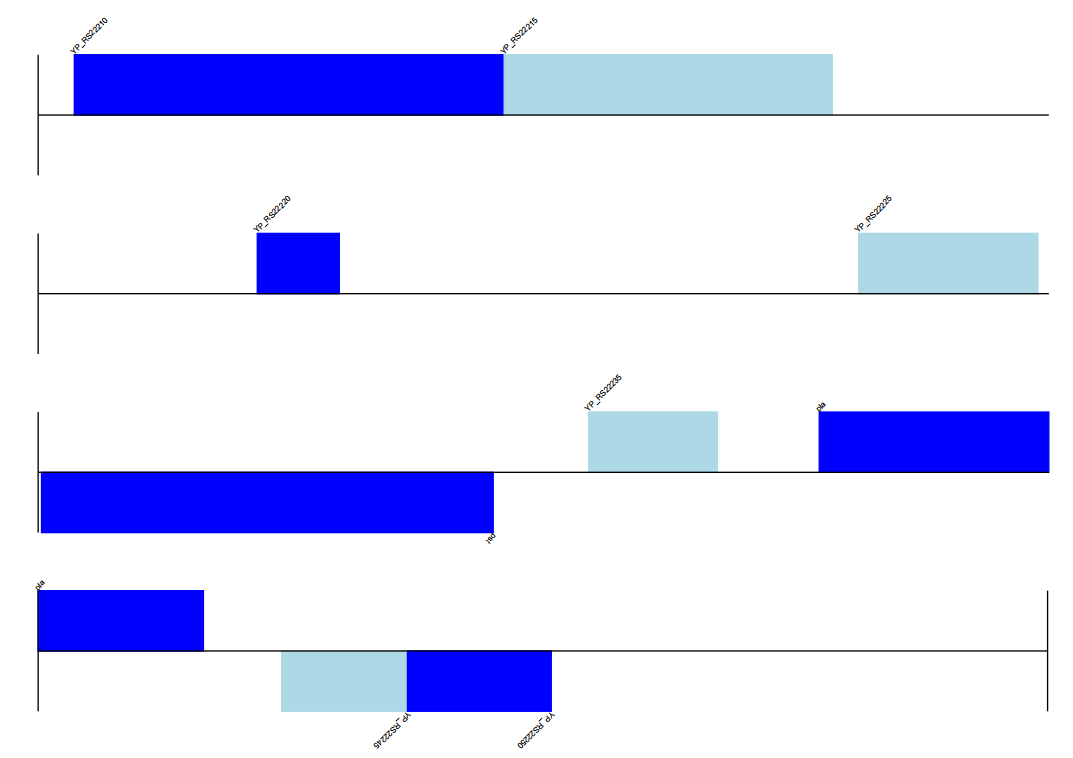
最后进行绘图即可,代码如下
>>> gd_diagram.draw(format="linear", orientation="landscape", pagesize='A4',
... fragments=4, start=0, end=len(record))
>>>
>>> gd_diagram.write("plasmid_linear.pdf", "PDF")
输出结果如下
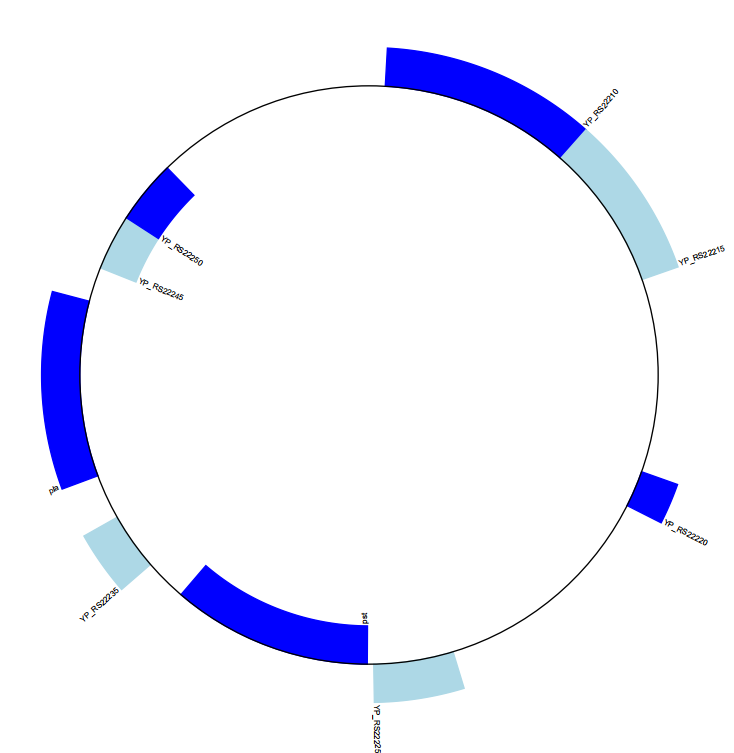
对于圈图,只需要修改简单修改绘图的参数即可,代码如下
>>> gd_diagram.draw(format="circular", circular=True, pagesize=(20*cm,20*cm),
... start=0, end=len(record), circle_core=0.7)
>>> gd_diagram.write("plasmid_circular.pdf", "PDF")
输出结果如下
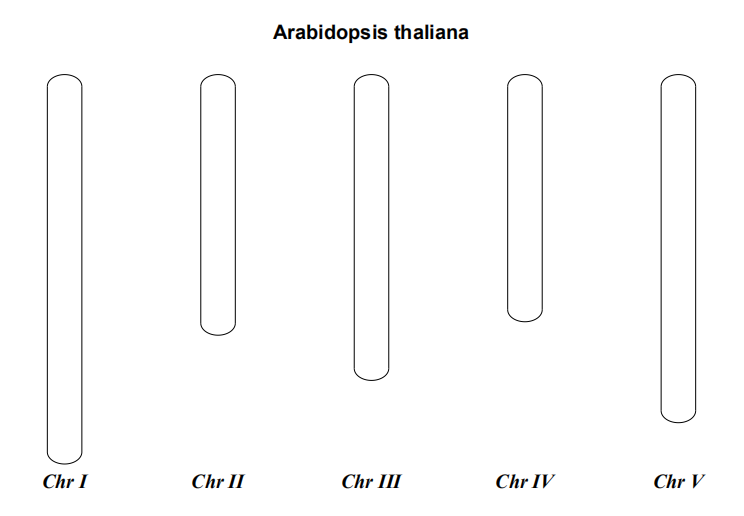
除了圈图之外,biopython还可以绘制染色体图。最简单的绘图,只需要提供染色体名称和对应的长度即可,代码如下
>>> entries = [("Chr I", 30432563),
... ("Chr II", 19705359),
... ("Chr III", 23470805),
... ("Chr IV", 18585042),
... ("Chr V", 26992728)]
>>>
>>> max_len = 30432563
>>> telomere_length = 1000000
>>>
>>> chr_diagram = BasicChromosome.Organism()
>>> chr_diagram.page_size = (29.7*cm, 21*cm) #A4 landscape
>>> for name, length in entries:
... cur_chromosome = BasicChromosome.Chromosome(name)
... cur_chromosome.scale_num = max_len + 2 * telomere_length
... start = BasicChromosome.TelomereSegment()
... start.scale = telomere_length
... cur_chromosome.add(start)
... body = BasicChromosome.ChromosomeSegment()
... body.scale = length
... cur_chromosome.add(body)
... end = BasicChromosome.TelomereSegment(inverted=True)
... end.scale = telomere_length
... cur_chromosome.add(end)
... chr_diagram.add(cur_chromosome)
...
>>> chr_diagram.draw("simple_chrom.pdf", "Arabidopsis thaliana")
输出结果如下
更进一步,可以在染色体上添加注释,标记基因组结构元件在染色体上的分布,代码如下
>>> chr_diagram = BasicChromosome.Organism()
>>> chr_diagram.page_size = (29.7 * cm, 21 * cm) # A4 landscape
>>>
>>> entries = [
... ("Chr I"
, "NC_003070.gbk"),
... ("Chr II", "NC_003071.gbk"),
... ("Chr III", "NC_003074.gbk"),
... ("Chr IV", "NC_003075.gbk"),
... ("Chr V", "NC_003076.gbk"),
... ]
>>>
>>> max_len = 30432563
>>> telomere_length = 1000000
>>>
>>> chr_diagram = BasicChromosome.Organism()
>>> chr_diagram.page_size = (29.7 * cm, 21 * cm)
>>> for index, (name, filename) in enumerate(entries):
... record = SeqIO.read(filename, "genbank")
... length = len(record)
... features = [f for f in record.features if f.type == "tRNA"]
... for f in features:
... f.qualifiers["color"] = [index + 2]
... cur_chromosome = BasicChromosome.Chromosome(name)
... cur_chromosome.scale_num = max_len + 2 * telomere_length
... start = BasicChromosome.TelomereSegment()
... start.scale = telomere_length
... cur_chromosome.add(start)
... body = BasicChromosome.AnnotatedChromosomeSegment(length, features)
... body.scale = length
... cur_chromosome.add(body)
... end = BasicChromosome.TelomereSegment(inverted=True)
... end.scale = telomere_length
... cur_chromosome.add(end)
... chr_diagram.add(cur_chromosome)
...
>>>
>>> chr_diagram.draw("tRNA_chrom.pdf", "Arabidopsis thaliana")
输出结果如下
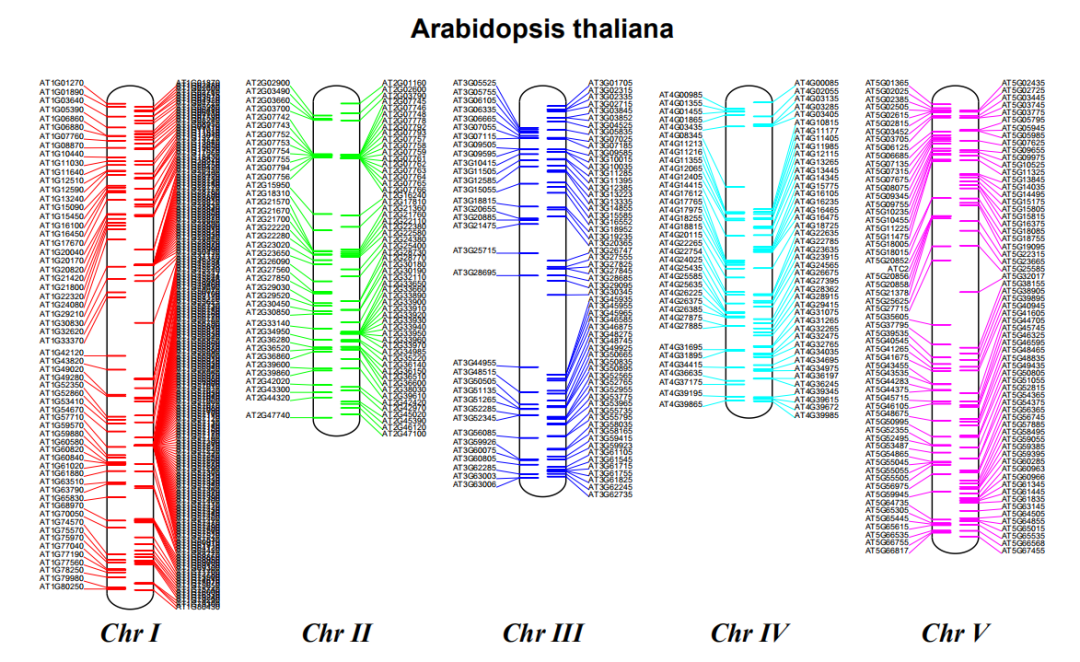
相比circos,biopython的track可能没有那么多种丰富的表现形式,但是也有其独特性。更重要的是,在染色体上标记特定元件的这种可视化方式,应用非常广泛,snp, ssr, cnv, genge等等都可以进行标记。
原创不易,欢迎收藏,点赞,转发!生信知识浩瀚如海,在生信学习的道路上,让我们一起并肩作战!
本公众号深耕耘生信领域多年,具有丰富的数据分析经验,致力于提供真正有价值的数据分析服务,擅长个性化分析,欢迎有需要的老师和同学前来咨询。转发本文至朋友圈,后台私信截图即可加入生信交流群,和小伙伴一起学习交流。扫描下方二维码,关注我们,解锁更多精彩内容!









