前面看到一个学妹在学习python,她的一篇推文中提到了一个python代码的学习资源
https://www.sc-best-practices.org/preamble.html
简介:
python单细胞学习:超详细的单细胞最佳实践,初学或者升级技能都合适
读《Single-cell best practices》2025实践版
python拟时序分析:推断成人人类骨髓的伪时间
今天看看里面的RNA速率分析:https://www.sc-best-practices.org/trajectories/rna_velocity.html
目的
轨迹推断(TI)领域的工具可以恢复在发育轨迹中的时间位置。但,传统的TI方法并不提供任何有方向的、动态的信息。此外,这些算法传统上不考虑转录组 reads 和相似性之外的信息。
细胞转录组特征的变化是由一系列事件触发的:大致来说,DNA被转录产生所谓的未剪接前体信使RNA(pre-mRNA)。未剪接的pre-mRNA包含与翻译相关的区域(外显子)以及非编码区域(内含子)。这些非编码区域会被剪接掉,从而形成剪接后的成熟mRNA。尽管scRNA-seq 协议无法在多个时间点捕捉转录组,但它们确实包含了区分未剪接和剪接mRNA读段的必要信息。
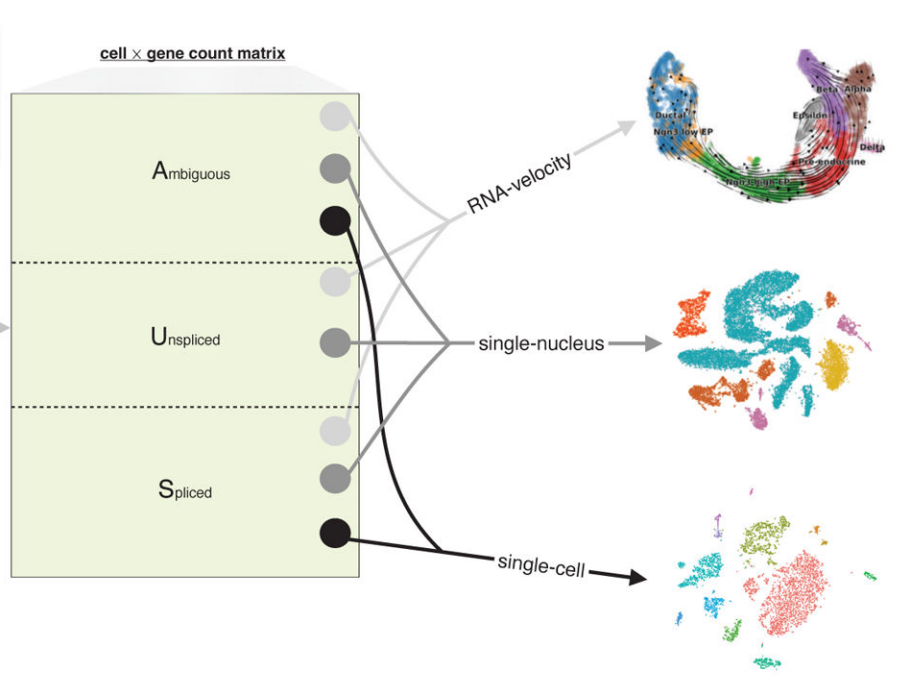
 RNA velocity of single cells. Nature, 560(7719):494–498, August 2018
RNA velocity of single cells. Nature, 560(7719):494–498, August 2018这篇2018年的Nature文章做出来的结果也太漂亮了:
模型理论:
识别未剪接和剪接的reads,可以构建一个描述剪接动力学的动态模型 ,并基于单细胞数据推断相应的模型权重。模型所描述的剪接RNA的变化被称为RNA速率 。当前的RNA速率模型假设的是基因特异性的模型。
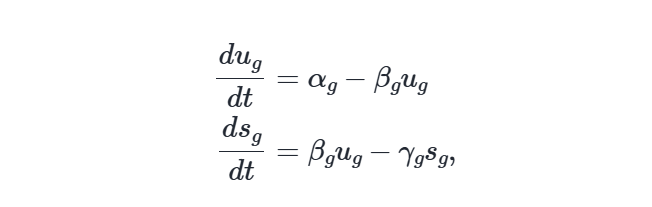
假设转录速率是 α,剪接速率是 β,以及剪接后RNA的降解速率是 γ。
参数推断
假设转录、剪接和降解的速率是恒定的,相平面图呈现出杏仁形状。上弧对应于诱导阶段,下弧对应于抑制阶段。然而,由于现实世界中的数据存在噪声,将未剪接的RNA计数与剪接的RNA计数进行对比时,并不能恢复出预期的杏仁形状。因此,数据需要先进行平滑处理。这个预处理步骤包括在细胞-细胞相似性图中,将每个细胞的基因表达平均化到其邻居细胞上。
稳态模型:
基本假设:
假设基因之间相互独立,且其底层动力学由上述模型控制。此外,还假设(1)动力学已达到平衡,(2)速率是恒定的,以及(3)所有基因存在一个单一的、共同的剪接速率。稳态本身位于相平面图的右上角(诱导阶段)及其原点(抑制阶段)。基于这些极端分位数,稳态模型通过线性回归拟合估计稳态比率。RNA速度则被定义为与该拟合的残差。
缺点:
尽管稳态模型能够在某些系统中成功恢复发育方向,但其本质上受到模型假设的限制。最容易被违反的两个假设是:所有基因存在一个共同的剪接速率,以及在实验过程中观察到平衡。因此,在这些情况下进行推断将得出错误的结果。此外,稳态模型仅考虑了数据的一个子集,且仅推断了稳态比率,而不是每个模型参数。
EM模型:
EM模型不再假设已经达到了稳态,也不再假设基因共享一个共同的剪接速率。此外,所有数据点都被用来推断剪接模型的完整参数集以及基因和细胞特异性的潜在时间。该算法使用期望最大化(EM)框架来估计参数。在E步中找到的未观测变量包括每个细胞的时间和状态(诱导、抑制或稳态)。所有其他模型参数在M步中被推断。
缺点:
尽管EM模型不再依赖于稳态模型的关键假设,因此适用范围更广,但推断出的RNA速度可能仍然违反先前的生物学知识。这种失败的原因主要有两个方面:一方面,EM模型仍然假设速率是恒定的。因此,每当这些假设不成立时,例如在红系成熟过程中,推断就是错误的。另一方面,该模型依赖于相平面图,就像它的前身一样。因此,当基因相平面图不遵循预期的形状时,该算法本质上是不适用的,并且会失败。
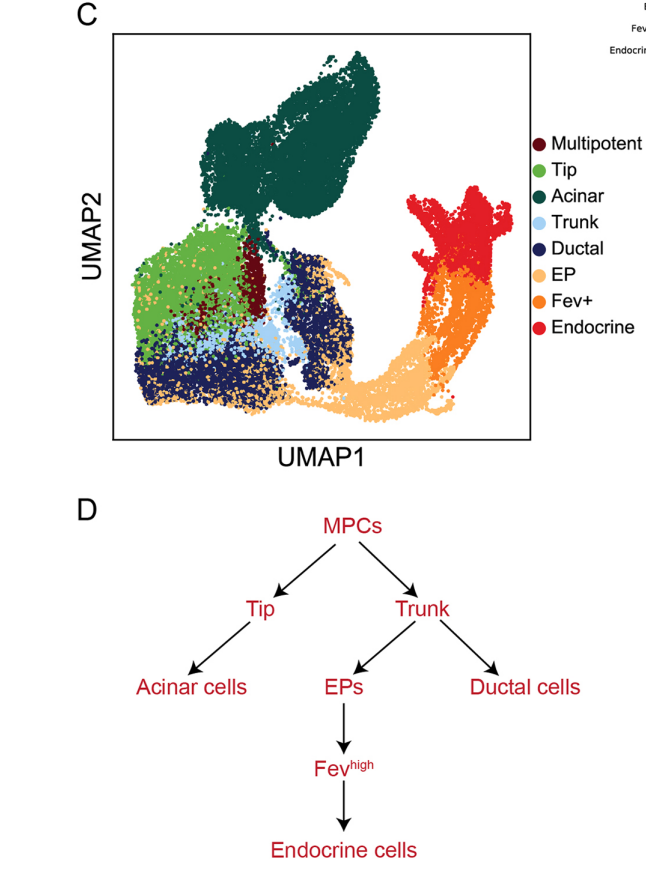
案例:胰腺内分泌生成中的RNA速度推断
本次分析胰腺的内分泌发育过程,数据来自文献《Massive single-cell mrna profiling reveals a detailed roadmap for pancreatic endocrinogenesis》。在这个系统中,前内分泌细胞(导管细胞、Ngn3低表达的内分泌前体细胞、Ngn3高表达的内分泌前体细胞、前内分泌细胞)会发育成四种内分泌细胞类型(α细胞、β细胞、δ细胞、ε细胞)。在这里,我们使用scVelo来推断RNA速度。
环境配置
bash命令安装:
## bash
conda activate sc
pip install -U scvelo -i https://mirrors.tuna.tsinghua.edu.cn/pypi/web/simple
导入库,下面开始是python代码:
from pathlib import Path
import scanpy as sc
import scvelo as scv
scv.settings.set_figure_params("scvelo")
DATA_DIR = Path("./data/")
DATA_DIR.mkdir(parents=True, exist_ok=True)
FILE_PATH = DATA_DIR / "pancreas.h5ad"
加载数据
内置在包中了:
adata = scv.datasets.pancreas(file_path=FILE_PATH)
adata
上面好像下载不下来,我找到了数据的链接:https://github.com/theislab/scvelo_notebooks/tree/master/data/Pancreas
adata = sc.read("endocrinogenesis_day15.h5ad")
adata
数据结构如下:
使用scVelo估计RNA速度,需要将未剪接和剪接的 counts 存储在AnnData的layers插槽中。建议将完整的counts,即未经处理的数据,传递给scVelo流程。
AnnData object with n_obs × n_vars = 3696 × 27998
obs: 'clusters_coarse', 'clusters', 'S_score', 'G2M_score'
var: 'highly_variable_genes'
uns: 'clusters_coarse_colors', 'clusters_colors', 'day_colors', 'neighbors', 'pca'
obsm: 'X_pca', 'X_umap'
layers: 'spliced', 'unspliced'
obsp: 'distances', 'connectivities'
数据预处理
由于scRNA-seq数据具有噪声且稀疏,因此必须对数据进行预处理,以便使用稳态模型或EM模型推断RNA速度。首先,筛选出未剪接和剪接RNA表达量均不足的基因(在此,至少为20)。随后,对未剪接和剪接RNA的细胞大小进行归一化处理,并对adata.X中的 counts 进行log1p转换,以降低异常值的影响。接下来,还识别并筛选出高变基因(在此为2000 )。
数据预处理与传统的scRNA-seq工作流程相似。在RNA速度的情况下,需要通过邻域的平均表达量来平滑观测值。这可以通过使用scVelo的moments函数来完成:
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
sc.tl.pca(adata)
sc.pp.neighbors(adata)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
在一个典型的工作流程中,需要对数据进行聚类,推断细胞类型,并将数据可视化为二维嵌入。这个示例的胰腺数据,这些信息已经预先计算好了,可以直接使用。
scv.pl.scatter(adata, basis="umap", color="clusters")
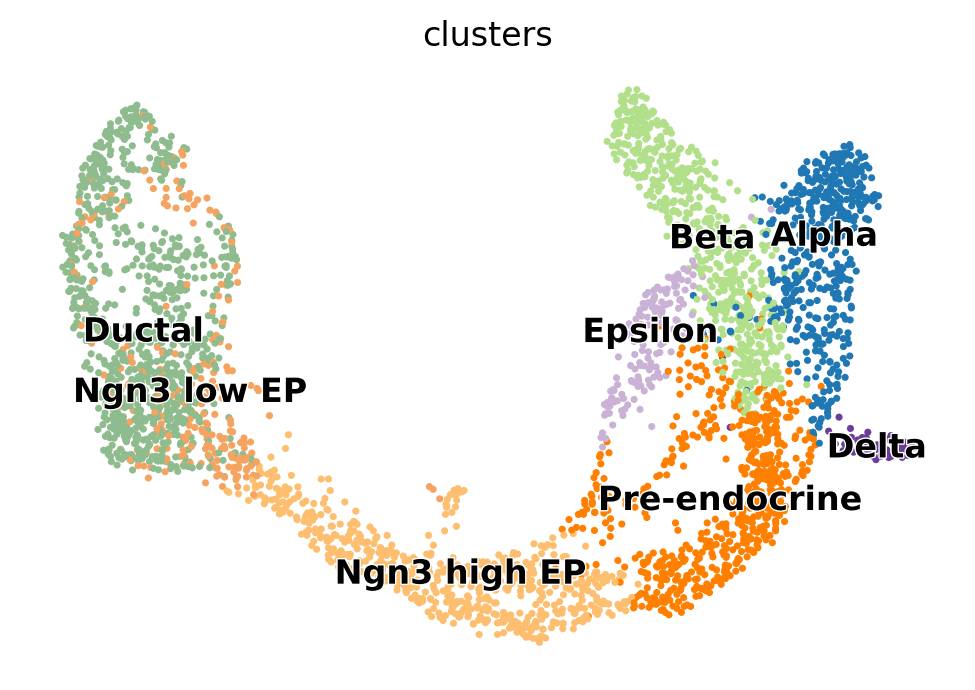
上面就是聚类且注释后的umap图。
RNA velocity推断:稳态模型
第一步,在稳态模型下计算RNA速度,调用scVelo的velocity函数,并将模式设置为“deterministic”:
scv.tl.velocity(adata, mode="deterministic")
scv.tl.velocity_graph(adata, n_jobs=1)
scv.pl.velocity_embedding_stream(adata, basis="umap", color="clusters")
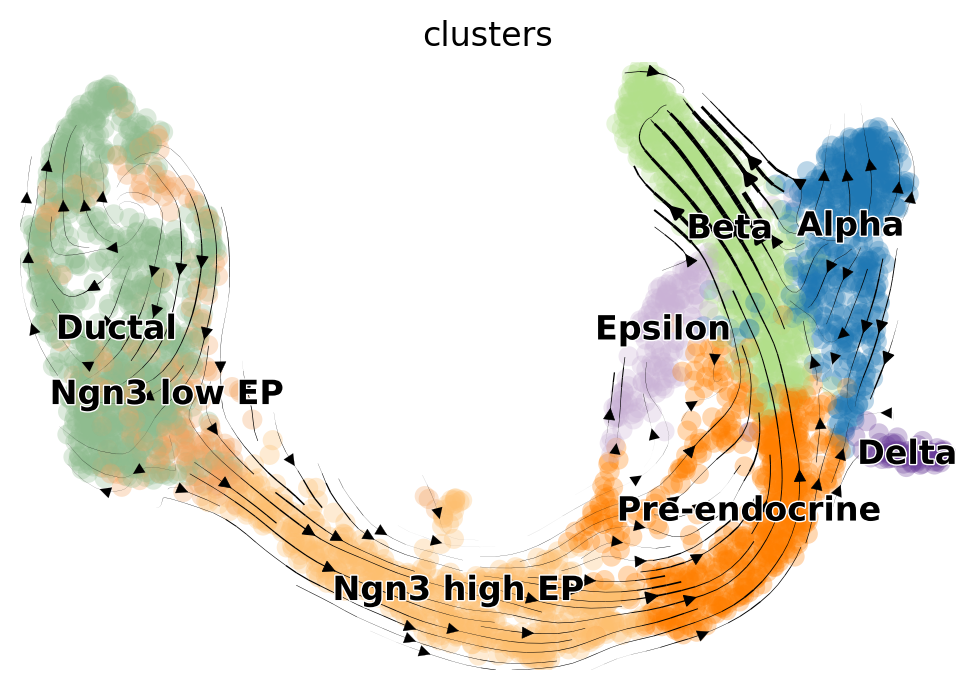
RNA velocity推断:EM模型
为了使用EM模型计算RNA速度,首先需要推断剪接动力学的参数。这一推断过程由scVelo的recover_dynamics函数来完成:
scv.tl.recover_dynamics(adata, n_jobs=1)
top_genes = adata.var["fit_likelihood"].sort_values(ascending=False).index
scv.pl.scatter(adata, basis=top_genes[:5], color="clusters", frameon=False)
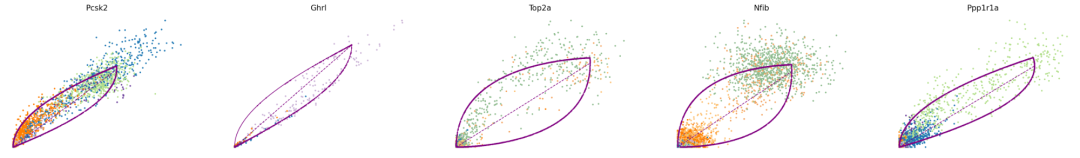
展示的五个基因中有三个(Pcsk2、Top2a、Ppp1r1a)的相平面图呈现出(部分)杏仁形状。
在估计了动力学速率(存储在adata.obs的fit_alpha、fit_beta、fit_gamma列中)之后,可以计算RNA速度以及其在二维UMAP嵌入上的投影。
scv.tl.velocity(adata, mode="dynamical")
scv.tl.velocity_graph(adata, n_jobs=1)
scv.pl.velocity_embedding_stream(adata, basis="umap")
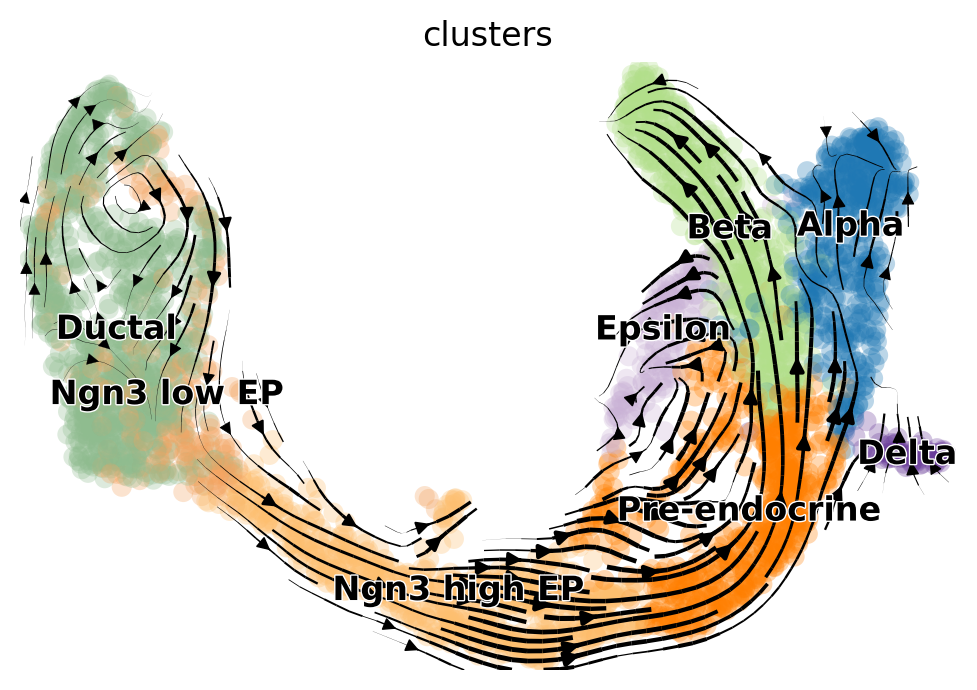
基于二维投影,EM模型更准确地捕捉了导管细胞中的细胞周期。此外,稳态模型的投影显示出从α细胞到前内分泌细胞的“逆流”。
小结
RNA速度分析是否适用于给定的数据集,需要注意以下几点:
所研究的发育过程的时间尺度必须与RNA分子的半衰期相当。例如,在胰腺内分泌生成中 满足这一要求,但在像阿尔茨海默病或帕金森病这样的长期疾病中则不满足。同样,RNA速度分析也不适用于稳态系统,例如外周血单核细胞,因为这些细胞缺乏(成熟)细胞类型之间的转换。
只有在底层模型假设(大致)成立的情况下,RNA速度才能被稳健且可靠地推断出来。为了验证这些假设,可以研究相平面图,以确认它们是否呈现出预期的杏仁形状。如果一个基因包含多个显著的动力学过程,那么在应用RNA速度分析时应格外谨慎,并且可能需要将数据细分为单独的谱系。
高维的RNA速度向量是通过将它们投影到数据的低维表示上来可视化的。然而,这种方法可能会导致错误和误导,因为投影的速度流高度依赖于(1)包含的基因数量和(2)选择的绘图参数。此外,投影质量在低维嵌入的边界处会降低。
今天分享到这~







