今天给大家介绍INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES的一篇文章:RPITER: A Hierarchical Deep Learning Frameworkb for ncRNA–Protein Interaction Prediction。文章提出了一个分层深度学习框架RPITER,在预测RNA-蛋白质相互作用(RPI)方面表现良好,并且可能优于以前的大多数方法。所提出的RPITER可以成为预测RPI和构建RPI网络的补充方法,将有助于推动ncRNA和lncRNA的相关生物学研究。
1.研究背景
非编码RNA(ncRNA)在多个基本生物学过程(如转录后基因调控)中起着至关重要的作用,并且与许多复杂的人类疾病有关。大多数ncRNA通过与相应的RNA结合蛋白相互作用起作用。对ncRNA-蛋白质相互作用的研究是理解ncRNA功能的关键。然而,目前用于鉴定RNA-蛋白质相互作用(RPI)的生物实验技术仍然昂贵且耗时。由于ncRNA-蛋白质相互作用的分子机制复杂,以及ncRNA(尤其是长ncRNA)缺乏保守性,ncRNA-蛋白质相互作用的预测仍然是一个挑战。基于深度学习的模型由于其强大的特征学习能力,已成为一系列生物序列分析问题中的最先进的模型。
2.数据
2.1 数据集
文章采用了以往研究中的5个基准数据集(表1)来验证所提出的方法。由于RPI369,RPI2241和NPInter数据集缺乏非相互作用对作为训练模型中的负样本,因此随机选取蛋白质和RNA片段,与阳性样本进行比对,去除相似性较高的相互作用对,从而得到对应的阴性样本库。表 1.本研究中使用的五个基准RPI数据集

2.2 数据预处理
要将RNA和蛋白质序列输入深度学习或传统机器学习模型,必须首先将序列数据转换为数值表示。由于文章数据集中的RNA和蛋白质序列长度在较大范围内(0-4000)变化,因此文章采用并改进了CTF方法,该方法按顺序计算k-mer频率以形成固定长度矢量表示。
在以前的CTF方法中,计算蛋白质的3-mer频率和RNA的4-mer频率以形成序列编码载体。对于蛋白质,20个氨基酸根据其偶极矩和侧链体积分为七组:{A,G,V},{I,L,F,P},{Y,M,T,S},{H,N,Q,W},{R,K},{D,E}和{C}。然后,每个蛋白质序列根据分组简化表示。因此,通过计算3-mer频率,蛋白质序列转换为包含343个元素的数字向量。对于RNA,使用四种核糖核苷酸(A,U,C,G),RNA序列用256个元素的数字向量表示。
在这项研究中,当仅考虑序列信息时,文章在蛋白质的k-mer频率编码过程中将k的范围扩展到1-3,并将RNA的k-mer频率编码过程中的k范围扩展到1-4。也就是说,对于蛋白质,文章计算了1-mer,2-mer和3-mer频率信息,形成一个包含399个元素的扩展编码向量。对于RNA,计算1-mer、2-mer、3-mer和4-mer频率信息,形成一个包含340个元素的扩展编码向量。通过将更多的序列信息添加到其编码向量中,文章希望增强模型输入,最终提高模型预测性能。这种序列编码方法称为改进型 CTF。
在考虑序列结构信息时,文章计算了蛋白质二级结构的1-3-mer频率和RNA二级结构的1-4-mer频率,以补充序列编码载体。对于蛋白质,结合三种二级结构的1-3-mer频率(α-helix, β-sheet and coil)与先前分为7组的简化序列特征组合,生成含有438个元素的蛋白质编码载体。对于RNA,将两种二级结构的1-4-mer频率与四个核糖核苷酸相整合,生成含有370个元素的核苷酸的RNA编码载体。该序列编码方法称为改进Struct-CTF。
3.模型设计
整个体系结构如图1 所示。文章的两种序列编码方法(改进的CTF编码和改进的Struct-CTF编码)用于两个序列编码部分。在每个编码部分之后,两个不同的网络架构(CNN和SAE)被采用在两个模块中,以从输入中提取特征并形成高级表示。在每个模块中,蛋白质和RNA编码载体分别由两个相似的部分进行分析,以形成各自的序列嵌入表示。最后,集成模块集成了四个基本模块(Conjoint-CNN,Conjoint-SAE,Conjoint-Struct-CNN和Conjoint-Struct-SAE)的输出,以形成RPITER的整个架构。

图 1.RPITER 的流程图
在模块Conjoint-CNN和Conjoint-Struct-CNN中,首先,使用CNN的两个相似的序列嵌入部分分别分析RNA和蛋白质输入载体并形成两个序列嵌入。每个带有CNN的序列嵌入部分都有三个卷积层,在两个卷积层之间,最大池化层用于降维并为噪声引入不变性。最后,一个三层全连接部分,第一个 Dense 层的输入是蛋白质和 RNA 嵌入的串联,最后一个 Dense 层的输出是预测结果,由后面的集成模块进一步集成。在模块Conjoint-SAE和Conjoint-Struct-SAE中,首先,使用SAE的两个相似的序列嵌入部分分别分析RNA和蛋白质输入载体并生成两个序列嵌入。每个带有SAE的序列嵌入部分都有三个完全连接的层,用于对序列信息的降维和高级特征提取。最后,三层全连接部分连接前两个序列嵌入作为其第一层的输入,并对第三层的特定RNA-蛋白质对进行相互作用预测。与之前的基于 CNN 的模块类似,两个基于 SAE 的模块的预测结果由后面的集成模块进一步集成。最后一个集合模块将前两个 CNN 模块和两个 SAE 模块的预测结果连接为其输入张量,并为给定的 RNA-蛋白质对生成更全面的预测结果。4. 结果
通过综合实验比较不同序列编码方法和RPI预测方法的性能。文章使用六个指标来比较方法性能,即准确度(Acc)、灵敏度(Sn)、特异性(Sp)、精密度(Pre)、马修斯相关系数(MCC)和AUC(受试者工作特征曲线下的面积(ROC))。4.1. 不同序列编码方法的性能比较
文章通过补充更多的主要序列信息和序列结构信息来改进序列编码方法CTF,称为改进的CTF和改进的Struct -CTF。表2和图2显示了五倍交叉验证(CV)中三种序列编码方法在数据集RPI2241上的性能。如图2 所示,三种方法的ROC曲线非常接近,但改良CTF(橙色)和改良Struct-CTF(绿色)的曲线略高于CTF(蓝色)的曲线。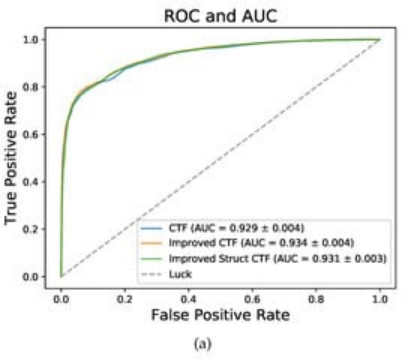
图 2. 通过五倍交叉验证,比较数据集RPI2241上不同序列编码方法的ROC和AUC
表 2.通过五次交叉验证,在数据集RPI2241上比较联合三元组特征(CTF)和文章的两种改进编码方法的性能

4.2. 不同基本预测模型的性能比较
RF和SVM被用来构建RPISeq的分类器;IPMiner将RNA和蛋白质特征输入到RF中以进行预测。文章提出的RPITER在四个基本预测模块中采用了CNN和SAE架构,以将输入特征映射到高级特征表示并生成预测结果。RPISeq、IPMiner 和 RPITER 都依赖于 CTF 表单序列编码功能。因此,文章比较了不同预测模型(即RF、SVM、CNN和SAE)的性能,在数据集RPI1807、RPI2241和NPInter上使用相同的CTF编码特征。表3列出了这些方法在数据集NPInter上的详细性能。表 3.通过五重交叉验证,对数据集NPInter上的卷积神经网络(CNN)、堆叠自动编码器(SAE)、随机森林(RF)和支持向量机(SVM)进行性能比较

在数据集NPInter上,CNN获得的Acc为0.953,分别比SAE、RF和SVM增加0.012、0.010和0.020。在数据集RPI1807上,CNN、SAE和RF之间的准确度差异很小,SVM的准确度最低。在数据集RPI2241上,SAE、RF和SVM的Acc分别为0.859、0.855和0.805,而CNN模型的Acc为0.887(比SAE和RF增加约0.03,比SVM增加0.08)。
4.3. RPITER不同模块的性能比较
文章的整个模型RPITER由四个基本的预测模块组成,即Conjoint-CNN
,Conjoint-SAE,Conjoint-Struct-CNN和Conjoint-Struct-SAE。文章的四个基本模块和整个架构 RPITER 在五个基准数据集上的预测精度为五倍 CV如图3所示。
图 3.RPITER不同预测模块之间的性能比较
使用五个基准数据集比较不同预测模块的性能,发现当训练样本足够时,基于CNN的模块比基于SAE的模块具有优势。另外,没有一个单独的模块可以始终超过所有数据集上的所有其他模块。文章提出的RPITER的架构结合了四个基本模块的优点,具有不同的架构和序列编码方法,可以提供更全面的RPI预测结果。
5. 总结
在这项研究中,文章提出了一个基于分层深度学习的框架RPITER,它包含两个序列编码部分作为输入,涉及两个基本的CNN和SAE网络架构来生成综合预测结果。设计的深度学习模型RPITER可以整合四个不同基本预测模块的优势,并提供全面的RPI预测结果。通过对5个基准数据集的实验,与之前的方法相比,RPITER在预测RPI方面表现出良好的性能。








