三、结果
1、用蛋白质编码基因表达训练的前馈神经网络区分正常组织和癌组织
为了揭示通常定义癌症状态的转录组学特征。对来自 GTEx 和 TCGA 的 11 对正常组织及肿瘤配对样本进行差异基因表达分析,然后查看失调基因中的overlap,结果表明仅有很少的蛋白质编码基因在六种或更多肿瘤类型中始终上调或下调[Fig.1a]。
为了克服对常见癌症转录组特征幼稚研究的局限性,作者试图训练能够区分正常和癌症样本的可解释深度学习模型。
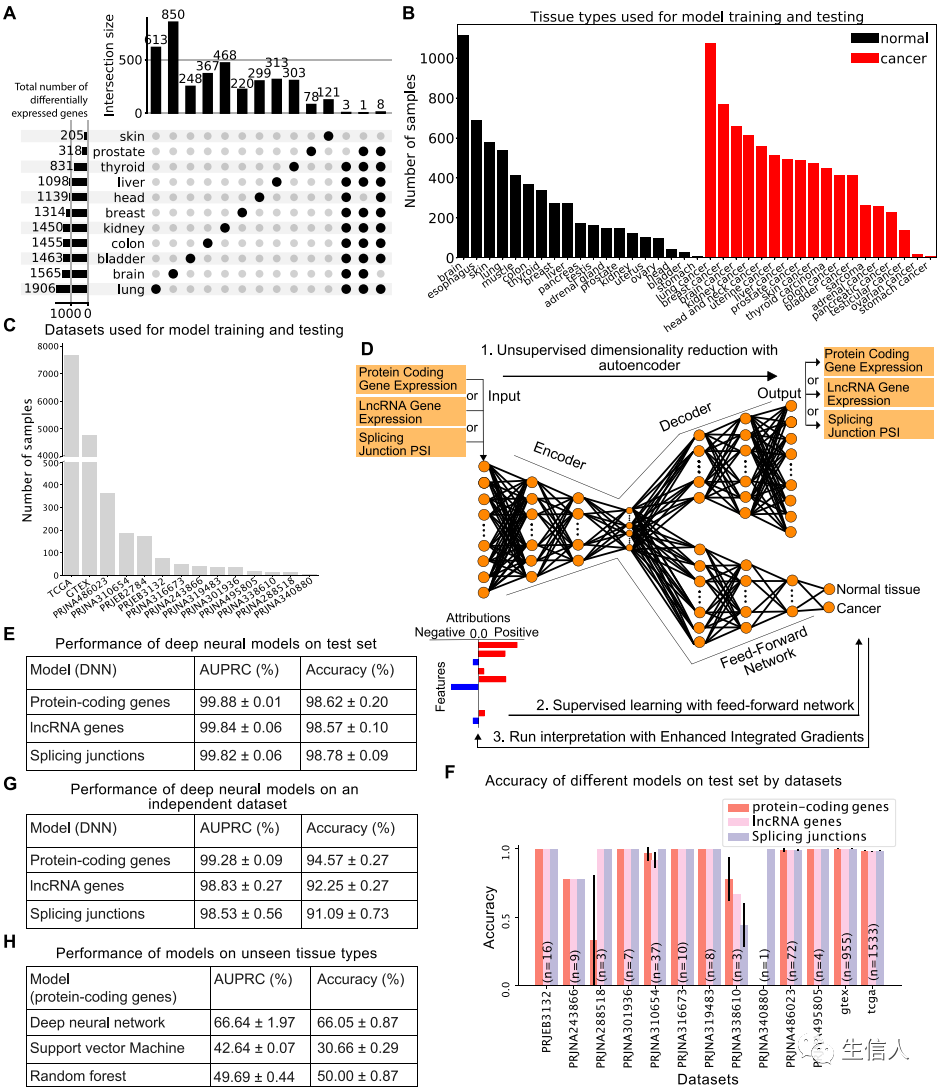
首先,整合一个大型RNA-Seq数据集,包含19种正常组织类型和18种肿瘤类型的13,461 个样本,并将数据分为反映癌症状态的两类:正常或肿瘤[Fig.1bc]。同时,使用12个较小的数据集来校正特定于数据集的偏差,这些数据集要么仅包含肿瘤样本,要么仅包含来自同一患者的肿瘤和配对的正常样本。作者还考虑了一个替代的方法——均值校正,例如常用的COMBAT方法,但这种方法严重限制了可用于模型训练的数据和基因集。
然后,使用来自19,657个蛋白质编码基因均值校正的表达数据,来训练一个自动编码器进行降维;使用有监督的深度神经网络来预测癌症状态。在验证集上调整模型超参数(学习率、隐藏层、节点数、激活函数和丢失概率),并使用验证集上性能最佳的超参数来修复模型架构。
最后,为了确保模型不会学习特定于数据集的偏差,使用一个额外的数据集评估该模型的效能。结果表明,蛋白质编码基因表达模型能够精确的预测样本来自正常组织还是肿瘤组织(Fig.1efg)。
为了评估该模型对于其他癌症类型的效能(训练集未包含该癌症类型),从三个额外的数据集中组织了一个新的数据集(包含正常细胞和恶性血液细胞);在不进行批次校正的情况下,评估深度学习模型表现。令人惊讶的是,尽管训练集和测试集数据之间存在显著差异,但该模型仍旧能够成功地将正常和癌症样本与血液区分开来(Fig.1h)。
另外,在相同的数据集中训练支持向量机和随机森林模型,作者发现,虽然在相同的独立数据集上进行测试,三个模型的效能相似。但当将支持向量机模型和随机森林模型用于血液数据集时,这两个模型完全失效(Fig.1h)。这表明,与常用的机器学习方法相比,深度神经网络模型更准确、更稳健。
2、lncRNA表达或剪接位点使用曲线足以定义癌症状态
其他类型的转录组特征,包括lncRNA表达和RNA剪接,已被用作预后标志物或预测癌症中的药物应答。同时,少量位于lncRNA基因中的突变或破坏蛋白质编码基因中的剪接已被证明会驱动癌症发生。但是,目前尚不清楚lncRNA表达或RNA剪接的广泛变化是否是癌症发生的广泛特征。作者尝试使用这些转录组学特征来区分正常组织和肿瘤组织。
使用相同的方法训练lncRNA模型和剪接点连接数据模型,值得注意的是,这些模型分别实现了 98.57% ± 0.1% 和 98.78% ± 0.09% 的准确度,具有高 AUPRC。正如蛋白质编码基因表达训练模型所观察到的那样,该模型在lncRNA基因表达数据上和剪接点使用训练模型上始终表现良好(Fig.1fg),这些结果进一步支持我们的模型的稳健性,因为它能够识别真正的生物信号而不是混杂因素。
3、深度学习网络的解释揭示了表征癌症状态的新转录组学特征
鉴于深度学习模型的高性能,作者想知道在我们的每个模型中,哪些转录组学特征是最重要的,以及这些特征是否主要由已知与癌症遗传相关的基因组成。为此,作者使用增强的积分梯度(EIG)生成了称为肿瘤样本归因值的特征重要性评分。
Ref:Enhanced integrated gradients: improving interpretability of deep learning models using splicing codes as a case study. Genome Biol. 2020
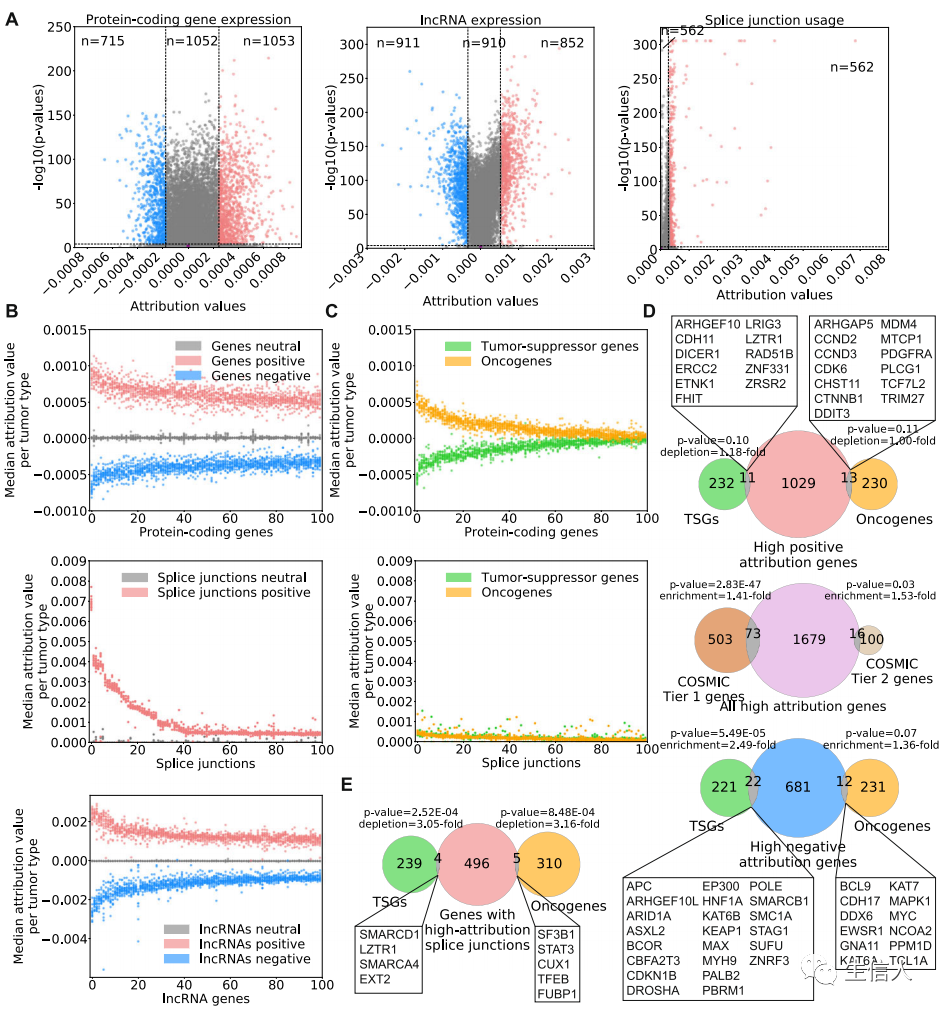
选择肿瘤类型中,具有较高归因值的蛋白质编码基因、lncRNA或剪接点,作为高归因值集合;同时选择归因值接近于零的的特征作为参考的Neutral集合(Fig.2a)。在14种肿瘤中探究癌症类型特异的归因值时,作者发现归因值前100的特征,在几乎所有肿瘤样本中都具有高的归因值(Fig.2b)。这表明,深度学习模型不是由样本量大的癌症类型中的异常表达或剪接点使用驱动,而是依赖于癌症的常见转录组学特征。
先前的差异分析表明,所有肿瘤类型中没有基因以相同的方式显著失调。与差异表达分析一致,作者发现给定基因的归因值的正负不一定反映癌症中基因表达的变化情况。也就是说,具有正归因值的基因不一定在大多数癌症中表达上调,或,具有高负归因值的基因不一定在大多数癌症中表达下调。因此,该模型的解释不是突出在许多癌症类型中相似变化的基因或剪接改变,而是暴露出癌症中始终偏离正常的转录组变异。
接下来,作者试图评估已知癌基因或抑癌基因与该模型归因值之间的关系。作者发现了一个显著的区别——癌基因获得正归因,而抑癌基因获得负归因值(Fig.2c)。然而,相对于模型中识别的该归因值特征,大多数癌基因或抑癌基因获得较低的归因值,甚至一部分归因值接近于0。作者只观察到一小部分高负归因值的基因在COSMIC基因中富集(Fig.2de)。
4、表征癌症状态的转录组特征的遗传改变频率
接下来,作者想知道高归因基因中以前未报告的遗传改变是否可能推动模型强调的转录组变异。作者在TCGA样本中证实了高归因值基因几乎不携带驱动突变(driver mutations)(Fig.3a)。但分析表明,具有高负归因值的基因的样本展现了更高的乘客突变频率,相比于Neutral集合来说(Fig.3b)。同时,结构变异的频率虽然在高归因基因中高于其参考的Neutral集合,但在所有高归因基因组中都低于COSMIC基因(Fig.3c)。同样,高归因基因受扩增或缺失事件影响的频率与Neutral集合或 COSMIC 基因没有显着差异(Fig.3de)。
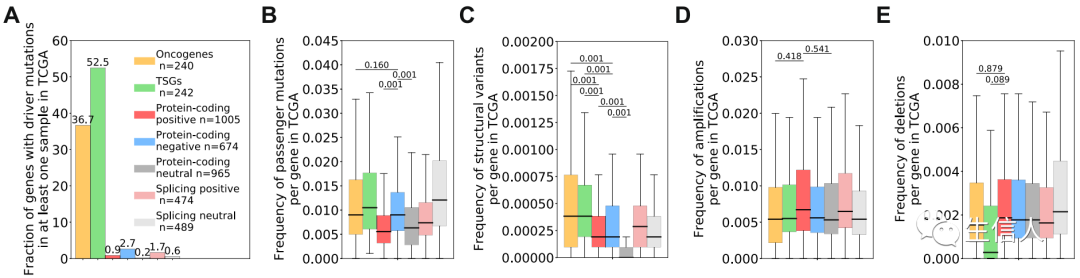
总体而言,深度学习模型确定的癌症转录组学特征并不经常受到遗传改变的影响,这表明从模型中获得的癌症表达和剪接模式不是由这些基因的遗传变异驱动的。
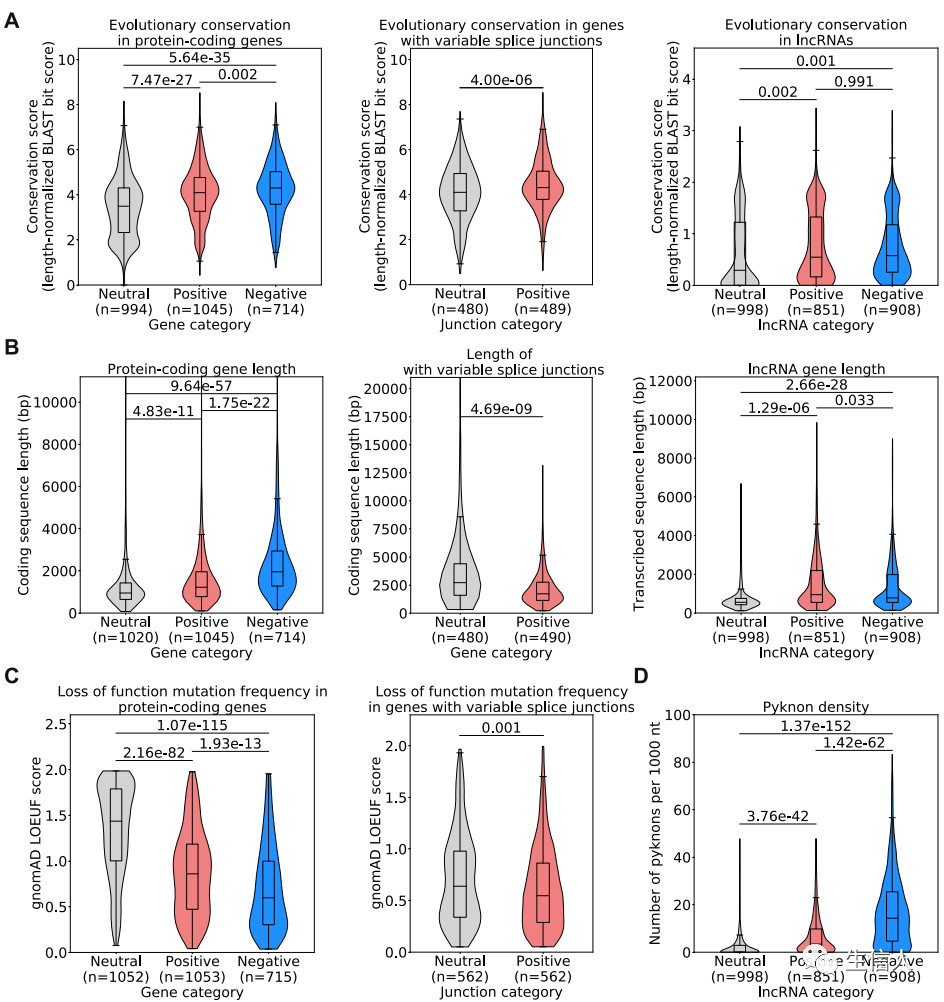
5、定义肿瘤状态的转录组学特征的高度进化和选择性限制
在通过表达或剪接连接使用建立了具有高归因值的基因列表,并发现这些基因中的大多数与COSMIC癌基因或抑癌基因不对应之后,作者试图探究深度学习模型中具有高归因值的转录组学特征,是否具有表明细胞中重要作用的特性。
作者发现,和Neutral集合相比,模型中具有高归因值的蛋白质编码基因,lncRNA基因和相对应的剪接连接的基因具有高度的进化保守性(Fig.4a)。同时,相对于参考的Neutral集合,具有高负归因的蛋白质编码基因以及正归因值负归因值的lncRNA显著的更长,但高归因值的剪接连接的基因显著的更短(Fig.4b)。具有高归因值的蛋白质编码基因和剪接连接基因对功能丧失突变展现了更高的选择性压力(Fig.4c)。最后通过pyknons方法,发现高归因值的lncRNA基因携带比Neutral集合更高的pyknons密度(Fig.4d)。
6、具有高归因值的剪接连接的表征
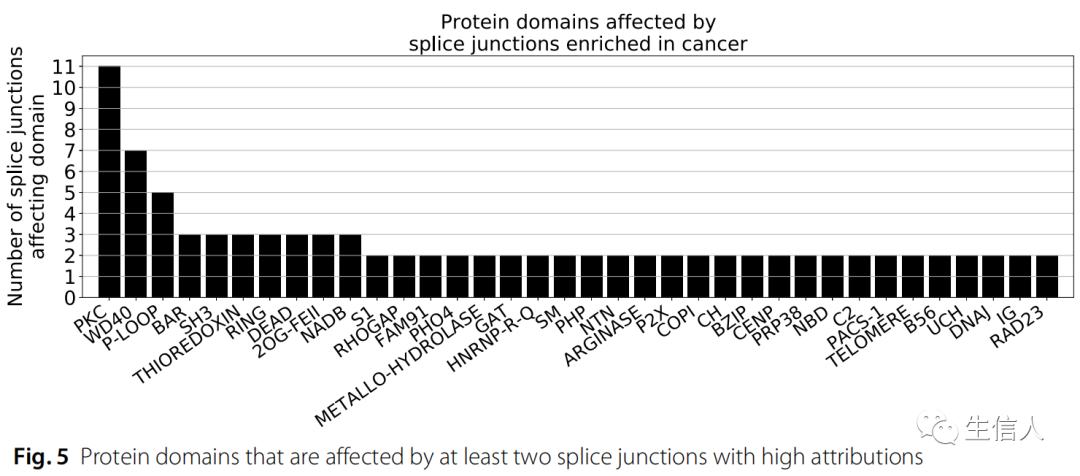
虽然很容易想象基因表达水平的变化如何驱动肿瘤发生,但解释剪接变化对疾病的影响并不那么简单。因此,作者试图预测具有高归因值的可变剪接连接如何影响蛋白质序列和功能。
作者首先注意到,高归因连接被预测会破坏基因的reading frame(阅读框架)。先前的研究表明,替代剪接可以通过靶向无序区域来调节蛋白质蛋白质相互作用。因此,作者研究了与可变剪接连接上下游两个外显子相对应的肽序列的预测无序性,但发现预测的肽无序水平在高归因连接中与在集合中观察到的无差异。
然后,使用NCBI保守结构域数据库,通过预测从高归因连接上游和下游的两个外显子编码的蛋白质结构域,评估高归因剪接连接是否会影响已知的蛋白质结构域。有趣的是,10个基因中的11个剪接连接会影响蛋白激酶C样超家族结构域的一部分转录本匹配序列(Fig.5)。作者还发现了额外的高归因剪接点,它们影响与癌症信号传导相关的其他结构域。
7、在癌症中具有高正归因值或负归因值的基因的对比功能
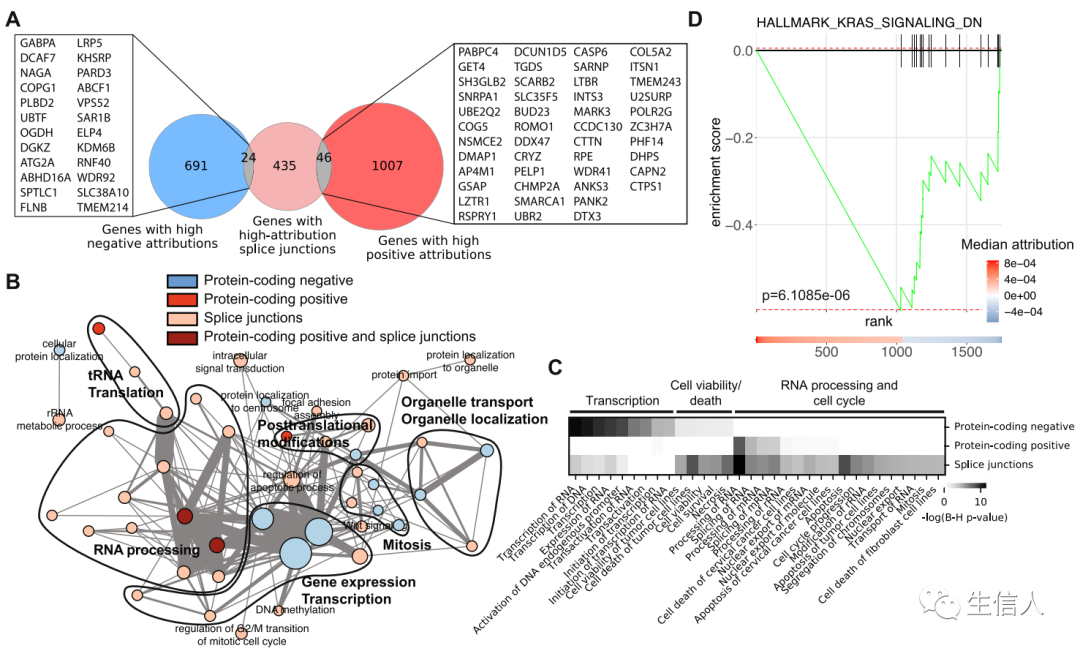
最后,鉴于模型中的大多数蛋白质编码基因或具有高归因值的剪接点的基因以前与癌症无关,作者试图了解这些基因的功能。首先,作者发现,通过表达识别的具有高归因值的基因与通过剪接点使用具有高归因值的基因,存在很大的差异(Fig.6a)。
对具有高归因值的蛋白质编码基因进行GO分析,发现具有高负归因值的蛋白质编码基因富含与转录、有丝分裂、组蛋白修饰、染色质调节和定位到中心体相关的功能,符合传统癌症观点。而具有高正归因值的蛋白质编码基因在转录后和翻译后修饰方面富集。同时,具有高归因值的剪接连接点的基因也富含与RNA加工相关的功能。另一方面,与生物学过程(BP)相关的富集图显示,通过表达或剪接的高正归因基因形成高度互连的网络,其核心与与 RNA 生物学相关的功能有关(Fig.6b)。与高归因基因相关的分子和细胞功能的Ingenuity Pathway分析证实,高负归因基因的功能与高正归因基因的功能不同,转录和RNA加工在两组中分别占据主导地位(Fig.6c)。