
材料计算模拟已经成为材料研究的重要组成部分。然而,现有的材料计算模拟方法很难兼顾计算速度与准确度:计算速度较快的方法得到的结果往往与实验测量有较大的误差,而计算误差较小的方法往往速度较慢,以至于无法大规模应用。因此,现有的大型计算材料数据库中的数据,经常与实验测量数据之间存在不可忽视的误差。为了缓解这一问题,近年来许多学者提出了各种不同的对于计算数据的校正方案,但是这些现有的校正方案一方面仍存在较大误差,另一方面往往只能用于特定材料体系,很难作用于所有材料。
为了克服这一问题,美国麻省理工学院材料科学与工程系Jeffrey Grossman课题组提出使用机器学习方法来校正密度泛函理论(DFT)的计算数据。在这项工作中,作者以材料形成焓为例,研究迁移学习(transfer learning)和多精度机器学习(multifidelity learning)对于校正材料形成焓预测的效果。作者发现,机器学习方法校正后的形成焓预测数据(基于PBE泛函)与实验数据的误差仅为~0.06 eV/atom, 显著低于传统修正方法修正后的GGA(PBE)泛函的计算数据(~0.1 eV/atom)和meta-GGA泛函的计算数据(0.08 ~ 0.1 eV/atom)。以此为基础,作者使用机器学习方法校正了Materials Project(MP)数据库中所有基于PBE泛函的形成焓预测,重新审视了MP数据库中记录的材料的稳定性预测,并发现了一系列在MP 数据库中稳定性可能被低估了的材料。此外,利用机器学习的方法,作者也揭示了DFT(PBE泛函)预测形成焓时误差出现的规律。此工作以标题“Calibrating DFT formation enthalpy calculations by multifidelity machine learning”发表在JACS Au上,并入选ACS Editors’ Choice. (https://pubs.acs.org/doi/10.1021/jacsau.2c00235)

图一:本文中使用的机器学习策略,即迁移学习和多精度机器学习。
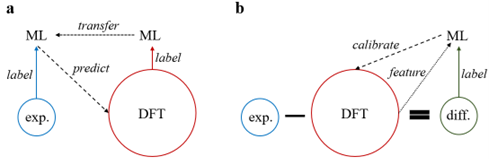
本文研究两种机器学习策略,迁移学习(图一a)和多精度机器学习(图一b),对于校正DFT (PBE)形成焓预测的效果。对于迁移学习,作者首先用DFT数据集训练神经网络,再把训练好的神经网络迁移以初始化一个新的神经网络,并用实验数据集训练被迁移的神经网络,最后用最终的神经网络预测材料的“实验“形成焓。对于多精度机器学习,作者首先建立实验与DFT计算之间的差值数据集,用差值来训练机器学习模型。在训练中,DFT计算数据亦可作为输入特征之一参与训练。训练完成后,机器学模型可以预测材料形成焓的实验与DFT计算数据之间的差值,进而实现对DFT计算数据的校正。本文用到了大约1000个实验形成焓数据,更多有关数据集的信息请参考原文。
图二:不同机器学习模型之间的比较。
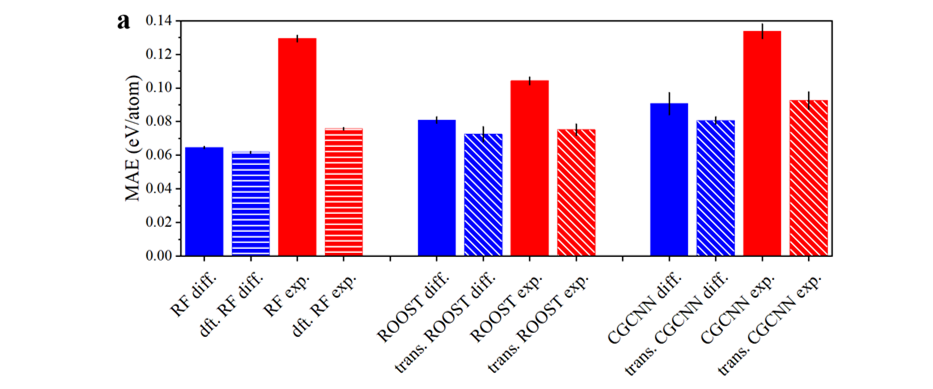
本文主要使用三种模型来实现迁移学习与多精度机器学习:随机森林(RF),ROOST和CGCNN。随机森林是基于材料描述符的经典机器学习方法,ROOST是基于材料的成分的深度学习方法,而CGCNN是基于材料成分与结构的深度学习方法。基于这三种模型,作者测试了一系列策略:直接学习实验值(exp.),学习差值(diff.),把DFT数据作为特征输入模型(dft.),以及迁移学习(trans.)。根据图二,作者发现,对于现有的形成焓数据集而言,最优的模型与策略是使用随机森林模型来学习实验与DFT之间的差值,同时把DFT数据作为输入特征。与MP数据库提供的形成焓预测值相比(基于PBE泛函的线性修正),本文中的最优模型可降低大约40%误差。(0.06 eV/atom vs. 0.10 eV/atom)。更多有关机器学习方法的讨论请参见原文。
图三:重新评估材料稳定性。
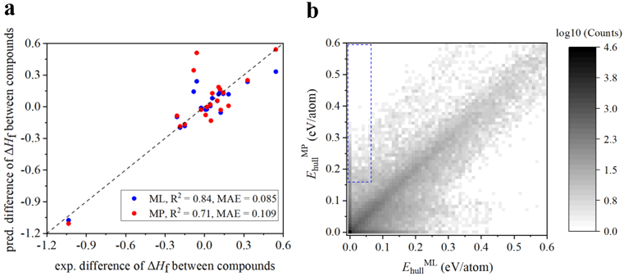
基于更精确的形成焓预测,作者希望重新评估材料的稳定性。首先,作者需要验证,更精确的形成焓预测是否能够更准确地判断材料的相对稳定性。如图三a所示,作者比较机器学习校正的形成焓与MP数据库中的形成焓,对于计算材料之间的相对形成焓的误差。作者发现,机器学习校正的形成焓能够更加准确地比较两个材料之间的相对稳定性。基于此,作者利用机器学习校正的形成焓重新计算了MP数据库中所有材料的energy above hull。如图三b左上角所示,作者发现有约800个材料在MP提供的energy above hull下不稳定,而在机器学习校正的形成焓导出的energy above hull下稳定。在这800个材料中,有将近100个材料已经被实验所合成。一个典型的例子是Mn-Sn合金体系。在MP数据库中,Mn-Sn之间没有稳定的金属间化合物,而实验上有一系列的稳定金属间化合物,例如Mn3Sn, Mn3Sn2 和 MnSn2。根据本文提供的机器学习模型,这些金属间化合物是稳定的,从而体现了本文的应用价值之一,既更准确的形成焓预测可以更好地判断材料稳定性。
图四:材料特征对于校正结果的影响。
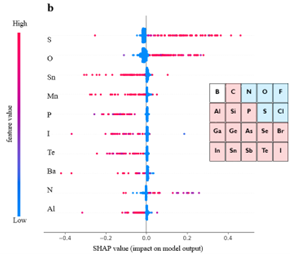
在校正形成焓的同时,机器学习模型也可以用来揭示DFT(PBE) 计算数值偏离实验测量值的规律。如图四所示,作者发现,材料中S, O, N含量越高,DFT倾向于低估(more negative,more stable)材料的形成焓,而材料中Sn, Mn, P, I, Te, Ba, Al的含量越高,DFT倾向于高估材料的形成焓。对于元素周期表右侧的非金属元素而言,右上角的元素往往使DFT低估材料的形成焓,而左下角的元素往往使DFT高估材料的形成焓。更多关于DFT偏离实验值的趋势的讨论请参考原文。









