全局机器学习力场(MLFF)能够捕捉分子系统中的集体相互作用,由于模型复杂性随系统规模显著增长,现在可以扩展到几十个原子。对于较大的分子,引入了局域性假设,因此未描述非局域相互作用。
在此,来自德国柏林工业大学的研究团队开发了一种精确的迭代方法来训练数百个原子的全局对称梯度域机器学习 (sGDML) 力场 (FF),而无需求助于任何潜在的不受控制的近似值。所有原子自由度在全局 sGDML FF 中保持相关,允许准确描述复杂的分子和材料,这些分子和材料呈现具有深远特征相关长度的现象。
研究人员在新开发的包含 42 到 370 个原子的分子的 MD22 基准数据集上评估 sGDML 的准确性和效率。该方法的稳健性在 MD22 数据集的超分子复合物的纳秒级路径积分分子动力学模拟中得到了证明。
该研究以「Accurate global machine learning force fields for molecules with hundreds of atoms」为题,于 2023 年 1 月 11 日发布在《Science Advances》上。

机器学习力场的优势与挑战
现代机器学习力场弥补了高效但极其近似的经典力场 (FF) 与昂贵的高级从头计算方法之间的精度差距。这种乐观基于机器学习 (ML) 模型的普遍性质,与经典力场 FF 中的静态参数化交互相比,这赋予了它们几乎不受限制的描述能力。
在构建模型时,传统的 ML 方法力求对手头问题做出一般假设,例如连续性和可微性。原则上,数据集中捕获的任何感兴趣的物理特性都可以通过这种方式进行参数化,包括过于复杂而无法从多体波函数中提取的集体相互作用。因此,MLFF 可以对量子多体机制提供前所未有的见解。尽管如此,与经典 FF 相比,全局 MLFF 的非凡表现力伴随着参数复杂性的显著增加。
作为权衡,许多 ML 模型重新引入了一些对原子间相互作用的经典力学限制。目前尚不清楚这种与无偏 ML 模型的背离在多大程度上损害了它们相对于经典 FF 的优势。
最近的几个 MLFF 模型引入了针对特定长程效应(例如静电)的经验校正项,但长程电子相关效应的特征仍然很差。物理交互模型增强的可用 MLFF 方法的数量表明,我们正在观察一个尚未确定通用解决方案的新兴领域。
全局模型的局限性
相比之下,全局模型能够包括所有相互作用尺度,但它们面临着必须耦合至少一组二次原子相互作用的挑战。这种缩放行为提供了严格的计算约束,减缓了近年来全局模型的发展。因此,当前全局模型仅限于只有几十个原子的系统大小,尽管准确的从头计算参考数据可用于更大的系统。
图 1:目前的全局 MLFF 只能扩展到几十个原子的系统大小,受到必须耦合二次原子相互作用的计算挑战的限制。(来源:论文)训练大分子的全局机器学习力场模型
在这里,研究人员开发了一种组合的封闭形式和迭代方法来训练大分子的全局 MLFF 内核模型。对这些模型的光谱分析表明,与 N^2 相比,大分子中的有效自由度数量显著减少,并且可以使用低维表示法捕获。利用这种洞察力,提出的大规模框架同时降低了模型的内存和计算时间要求。在两步过程中,有效自由度以封闭形式求解,然后迭代地将剩余的波动收敛到完整问题的精确解。
图 2:正则化 sGDML 核矩阵 Kλ(丙二醛,σ=12,λ=10^−10)在预处理 P^−1 Kλ 前后随诱导点数 k(占训练点总数 m 的 2.5%、5% 和 7.5%)增加的特征值谱。(来源:论文)该研究的重点是基于高斯过程 (GP) 的 ML 模型,因为它们具有几个独特的属性,例如线性和损失函数凸性,可以用来实现研究目标。
研究人员证明了其解决方案在对称梯度域 ML (sGDML) FF 上的有效性。它使我们能够可靠地为比以前更大的分子和材料重建 sGDML FF。
提出的训练方案可以处理包含数百个原子的系统,所有这些原子都在模型中完全耦合。使我们能够在稳定的纳秒长分子动力学 (MD) 模拟中研究超分子复合物、纳米结构和四大类生物分子系统。
大规模分子 FF 的评估
收敛性
为了评估方法的有效性,研究人员研究了预处理器配置对 CG 求解器收敛行为的作用。特别是,评估预处理器强度对整体训练成本的影响。该测试的目的是检查在实践中如何有效地解除封闭形式求解器的关键内存限制。
从 MD17 数据集(https://www.science.org/doi/10.1126/sciadv.1603015)中的丙二醛轨迹中代表性地采样了 1000 个点,这些点是通过增加预处理强度从 2.5% 到 100% 进行训练的。对于每个配置,都会测量求解器的运行时间和内存使用情况。
图 3:训练丙二醛 sGDML 模型所需的迭代次数(在 500 K 下采样,1000 个训练点,σ=12,目标残差低于 10^−4)作为预处理器强度(诱导点数)的函数。(来源:论文)分析表明,当使用超过 2.5% 左右的列来构建预处理器时,CG 求解器以一致的可靠性收敛。在此配置中,内存占用非常低,但运行时间超过了封闭式求解器。但是,如果使用超过 10%,则迭代训练方法的内存和运行时复杂性都会降低。这意味着较小的预调节器不仅构建和存储成本更低,而且实际上整体性能更好。
可扩展性
生成大量参考数据集的成本超过了 ML 模型可以提供的任何速度。在大尺度上,原子相互作用可能变得更加复杂,因为它们涉及更广泛的长度尺度。正是在这种情况下,真正需要 ML 模型的组合可扩展性和数据效率。
为了对迭代 sGDML 求解器进行最终测试,生成了一组新的 MD 轨迹 (MD22),涵盖多达数百个原子的系统。MD22 包括四大类生物分子和超分子的示例,从具有 42 个原子的小肽一直到具有 370 个原子的双壁纳米管。
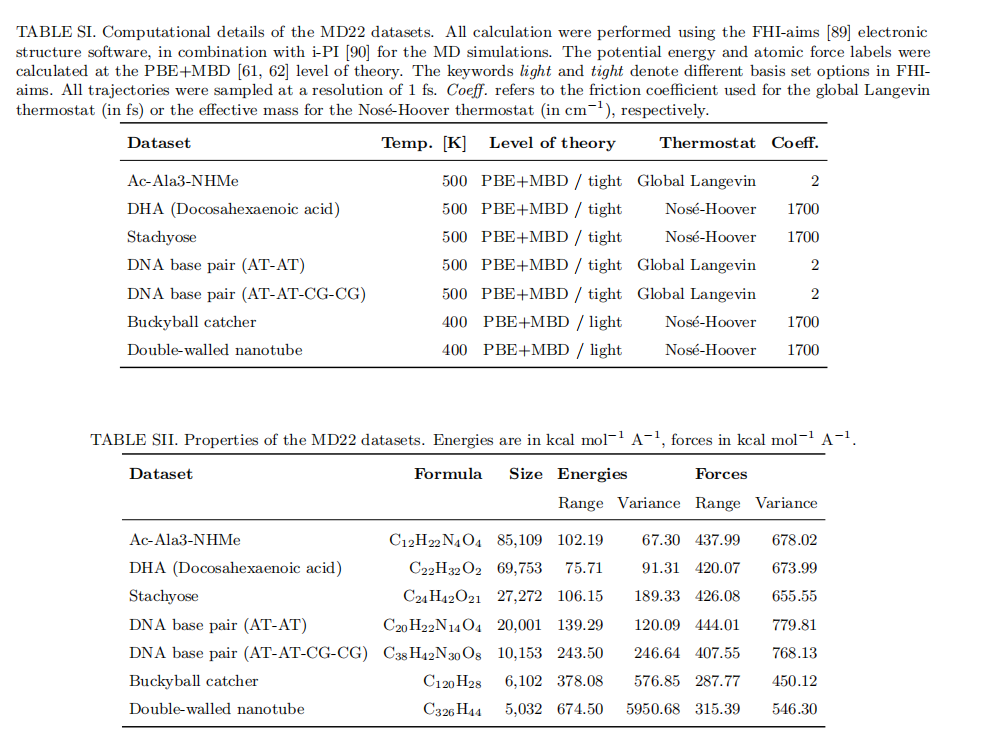
图 4:MD22 数据集属性。(来源:论文)
非局部交互的表示
为了研究经过训练的 sGDML 模型是否提供具有化学意义的预测,研究人员将 sGDML 应用于供体-桥-受体(DBA)型分子,该分子由两个苯环组成,这些苯环通过 E-乙烯部分连接形成共轭 π 系统(桥)。
一个具有化学意义的模型应该预测这种能量变化在整个 π 系统上是离域的(而不是通过旋转中心附近的局部变化来解释它)。为了定性地了解如何在 sGDML 模型中处理这些相互作用,研究了单个原子如何对预测做出贡献。作为原子之间成对相关性的线性组合,模型的部分评估揭示了原子对每个预测的贡献。
图 5:sGDML FF 预测的供体-桥-受体型分子(4-二甲基氨基-4'-硝基芪)的能量贡献。(来源:论文)
可以观察到,系统中的所有原子都参与了用 sGDML 生成预测,这对于将能量划分为局部原子贡献的模型是不可能的。表明 sGDML 模型学会了在整个分子的环旋转时使能量变化离域,这符合化学直觉。
讨论与展望
在这项工作中,研究人员提出了一种迭代方案,使 sGDML 能够稳健地应用于更大的系统,而无需引入对原始模型的任何近似。这是通过数值预处理方案实现的,该方案显着减少了学习问题的条件数,从而实现了 CG 迭代的快速收敛。
有了这一进步,我们现在能够将基于内核的模型应用于以前只能由神经网络访问的大规模学习任务,同时继承 sGDML 的样本效率和准确性。现在,可以在长时间尺度的 MD 模拟中研究表现出具有深远特征相关长度现象的分子系统。
MLFF 开发的突破通常是由提供不断变化的挑战的基准数据集的创建驱动的。在这项工作中开发的 MD22 基准数据集现在为原子模型在分子大小和灵活性方面提出了新的挑战,这可以进一步推进类似于以前的量子力学计算数据集的新型 MLFF 架构的研究。
该技术开发允许未来两种 MLFF 开发方法之间的交叉利用:现在,基于内核的 MLFF 可以利用 GPU 上可用的大规模并行性和支持深度神经网络可扩展性的软件基础设施。另一方面,内核方法的建模原理激发了新架构的开发,例如使用自注意力的 transformer,并为过度限制性的局部假设铺平了道路。迭代求解器独家使用即时模型评估也代表了基于内核的 MLFF 通常训练方式的范式转变,这为进一步开发开辟了新途径。
未来的研究将进一步探索我们在物理科学的广泛应用领域中显著更好的缩放行为。
https://www.science.org/doi/10.1126/sciadv.adf0873
感兴趣的读者,可以添加小邦微信(zhiyaobang2020)加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位 或
姓名-学校-职务/研究方向。










