论文题目:
Macrocyclization of linear molecules by deep learning to facilitate macrocyclic drug candidates discovery
今天给大家介绍一篇发表在Nature Communications上的文章,利用深度学习实现线性分子的大环化,以促进大环药物候选物的发现。迄今为止,在药物开发领域中,深度学习的当前应用主要集中在药物样品小分子上,利用深度学习算法对线性分子进行大环化的实施仍然是一个未充分探讨的领域。这其中的原因复杂多样,但最相关的原因之一可能是由于这些线性分子长期未受充分开发和利用,导致可用于模型训练的大环分子数量相对较少。在本文中,作者开发了一种基于深度学习的大环候选药物设计方法Macformer。对于给定的带有环化位点标记的起始线性分子,Macformer可自动添加结构多样性、且与线性分子具有较好兼容性的环链片段,生成具有结构多样性和新颖性的大环化合物,来探索其大环类似物的化学空间。Macformer在ChEMBL内部和ZINC外部测试集中均表现出较好的性能,其实用性也在大环类JAK2抑制剂设计的研究中得到实验验证。
大环化合物通常指的是由12个或更多原子组成的环状小分子或肽,它们在新药物发现领域已经崭露头角。这一类化合物具备独特的理化性质,包括高分子量和丰富的氢键供体,使其在化学结构上超越了Lipinski的五法则的限制。与它们的线性对应物相比,大环化合物往往倾向于采用预先组织的受限构象,并与靶点建立更为广泛的相互作用。因此,它们具备表现出更强的结合亲和力、提高的选择性或卓越的药理特性的潜力。大环化合物已成功地被用作潜在的治疗药物,针对各种药物靶点,包括激酶、蛋白酶和G蛋白偶联受体。特别值得注意的是,由于其独特特性,大环化合物被认为是针对一些传统小分子药物难以应对的具有挑战性的蛋白质的特殊化合物,从而弥合了小分子药物和大生物药物之间的差距。例如,大环化合物在市场上主导着乙型肝炎病毒NS3/4A的抑制剂,该蛋白具有浅且暴露于溶剂的凹槽,对小分子结合提出了挑战。此外,已经报道大环化合物在调控具有大、平坦和动态表面的蛋白质-蛋白质相互作用方面具有优势
在药物研发领域,分子大环化一般具有如下优势:1)实现骨架跃迁,突破现有专利限制;2)增加分子刚性,从而降低与靶标结合的熵损失,提高结合亲和力;3)提供较大的表面积,有利于结合口袋较浅的、传统小分子难以靶向的“困难”靶标;4)改善分子理化/ADMET性质如血脑屏障BBB性质,并增强分子稳定性。
在从生物活性线性分子出发进行大环化合物的合理设计时,通常包括两个关键步骤。首先,需要添加与线性化合物相兼容的大环连接基团,从而形成大环化合物。其次,需要评估大环化合物与靶点的结合口袋之间的兼容性。对于第二步,已有的研究方法相对明确,许多在药物设计中常用的模拟方法,如构象优化和分子对接,可以协助进行这一过程。如果我们能够通过在第一步中添加结构多样的连接基团来生成具有化学多样性的大环化合物,那么在随后的靶标-化合物结合预测后获得新颖的大环化候选物的机会无疑会增加。
然而,在最初阶段线性化合物的大环化主要是依靠药物化学家的经验知识驱动的。尽管最终的结果通常被呈现出来,但科学文献中通常没有充分描述涉及的详细步骤。这种不透明且非标准化的过程对于经验不足的研究人员来说很难遵循,而且经验知识也不足以覆盖大环连接基团的广阔化学空间。
针对环链片段添加问题,本研究结合子结构对齐的随机SMILES数据扩增技术和深度学习框架Transformer开发了自动大环化模型Macformer。Macformer在大环药物设计中的创新性主要在于以下两个方面:
1.采用Transformer框架,对输入线性分子SMILES和输出大环分子SMILES之间的全局依赖关系进行建模,推断与起始线性分子具有较好的整体兼容性的环链片段;
2.通过添加多样性的环链片段生成具有结构多样性和新颖性的大环化合物,以有效探索大环类似物的化学空间,提高获得新颖高效大环骨架的概率
这篇文章提出了 一种基于深度学习的大环候选药物设计方法Macformer。对于给定的带有环化位点标记的起始线性分子,Macformer可自动添加结构多样性、且与线性分子具有较好兼容性的环链片段,生成具有结构多样性和新颖性的大环化合物,来探索其大环类似物的化学空间。
具体的框架如图一所示。从ChEMBL数据库中收集了1.8万个大环化合物,通过循环切断大环上的每两个可断裂单键,获得23万个非环-大环字符串对,并按8:1:1划分为训练集、验证集和测试集。此外,从ZINC数据库收集了486个有活性的大环化合物,按照同样的处理方式获得5551个非环-大环字符串对,作为外部测试集。为了进一步扩大可训练的数据量,采用子结构对齐的随机SMILES字符串进行数据扩增。同经典SMILES和随机SMILES相比,采用子结构约束的随机SMILES字符串将输入和输出字符串之间的差异最小化,这使得Macformer可以在输出序列中较好地重现起始非环片段,同时有利于模型在分子生成过程中将更多的注意力放在环链片段的推断上。在ChEMBL测试集和ZINC测试集上,Macformer使用波束搜索算法推断目标序列,并输出排名前10的序列。
为了比较Macformer方法与已报道方法的优势,在这项工作中构建了基于三维片段数据库搜索和片段连接的自动环化方法MacLS。起始线性分子三维构象通过两种方式获得,一是从分子本身直接生成(MacLS_self方法),二是从预先构建的大环三维结构中提取(MacLS_extra方法)。采用距离、二面角等几何参数对片段进行过滤后,通过计算环链片段和非环起始结构连接键原子之间的RMSD值,对环链片段进行排序,并获得10个候选大环化合物。
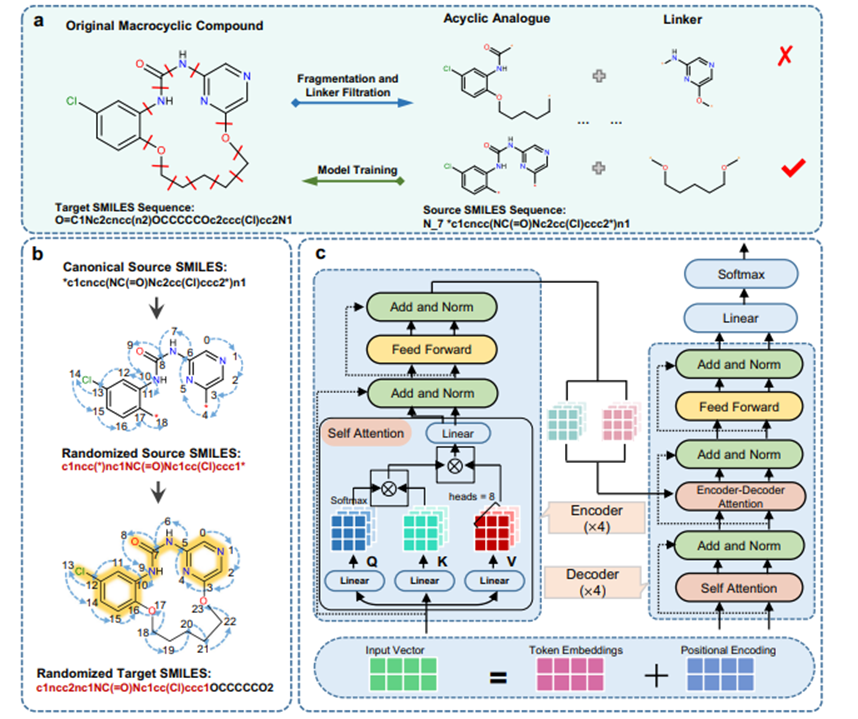
图 1. Macformer整体流程。a 数据预处理协议用于生成无环-大环SMILES对,以供模型训练和评估使用,“N_7”标记表示连接基团上最短路径的重原子数。b 使用随机化SMILES进行子结构对齐方式的无环-大环对数据增强。c Macformer的模型网络架构。
Macformer的性能首先在ChEMBL内部测试数据集上进行了评估,结果如表1所示。
与仅仅使用经典SMILES字符串的基线模型相比,进行数据扩增后,模型的所有指标均有提升,尤其是重现率(96.09 % vs 54.85%)和有效性(80.34% vs 66.74%)的提升最为明显。这表明使用子结构限制的随机SMILES字符串训练的模型不仅有利于重构大环骨架,而且有利于学习化学语言的基本句法表达,以产生具有化学意义并兼具结构多样性和新颖性的SMILES字符串。当非环构象为从头生成时,MacLS_self获得的大环分子的有效率仅为17.05%。当使用从大环分子三维结构中提取的构象时,MacLS_extra生成分子的有效率提高到89.65%,说明起始构象对片段库搜索方法非常重要。而Macformer方法由于使用SMILES序列,摆脱了对起始分子构象的依赖。此外,Macformer可以生成训练集中没有的新的大环链片段,在5倍数据扩增模式下,新颖环链片段占比为62.11%,而MacLS方法仅仅从预先构建的数据库中检索片段,因此新颖环链片段的占比为0%。此外,MacLS方法对目标大环分子的重构率比较低,MacLS_self给MacLS_extra方法的重构率仅为0%和4.16%。
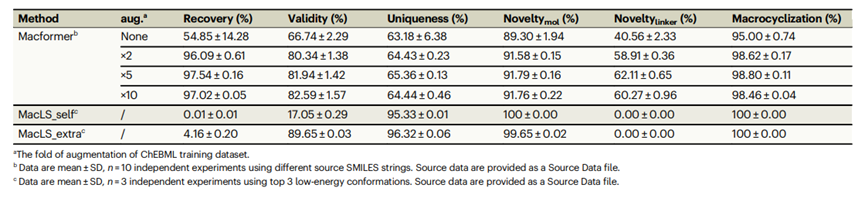
表 1.在ChEMBL测试数据集上,对Macformer不同数据增强次数以及MacLS的比较。
这些方法在另一个外部测试数据集上进行了进一步评估,该数据集包含5551对无环-大环SMILES,这些对是从ZINC数据库中的486个生物活性大环中提取的。与来自ChEMBL数据库的大环相比,这些大环具有更低的分子量和更短的SMILES长度。正如表2所示,增强型模型在外部ZINC数据集上也能够提供系统性改进的性能。经过5倍和10倍增强训练的两个模型均能恢复80%以上的原始大环化合物,生成84%以上的有效SMILES字符串,并实现99%以上的新颖性和大环化。这些结果表明,在数据增强情景下,Macformer表现出出色的泛化能力。
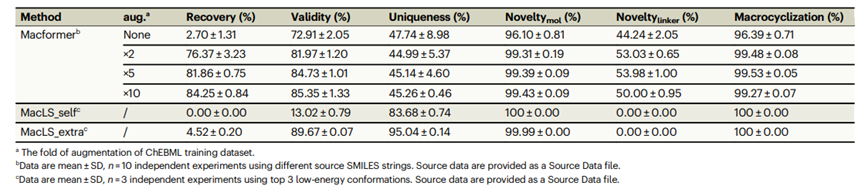
表 2. Macformer在不同增强数量下与MacLS在ZINC测试数据集上的比较
不考虑结构新颖性连接物的来源,Macformer和MacLS都表现出了生成具有结构新颖性的大环化合物的能力。这引发了一个问题,即这些新型化合物的化学空间是否存在差异。为了探讨这个问题,作者首先使用RDKit v2020.03.3.034中实现的Morgan指纹(2 bond radius)来评估生成的新型目标大环化合物与真实目标大环化合物之间的结构相似性。对于给定的目标化合物,计算其与所有相应生成的新型化合物的Tanimoto系数(Tc)值,然后求平均以得出最终分数。如图2a所示,由于无环化合物和大环化合物之间存在共同的亚结构,大多数生成的新型化合物具有平均Tc分数大于0.7。然而,Macformer倾向于生成与目标大环化合物更相似的新化合物,而MacLS_extra则不太相似。
上述结果有些出乎意料,因为Macformer可以推断出在训练数据集中不存在的新型连接物,而MacLS_extra则没有这种能力。因此,作者通过计算它们的1024位Morgan指纹来探索新型连接物的化学空间。此外,作者还利用均匀流形近似和投影(UMAP)算法进行降维。UMAP可以更好地保留原始高维空间中数据点之间的相似性关系,优于t-分布随机邻居嵌入。如图2b所示,Macformer在ChEMBL测试和ZINC数据集上生成的结构新颖连接物都位于环境中,这个环境包围着来自ChEMBL训练数据集的连接物。同时,除了执行大环化,Macformer还可以同时对起始线性亚结构进行微小修改,以生成与目标大环化合物具有高相似性的新结构(图2c)。
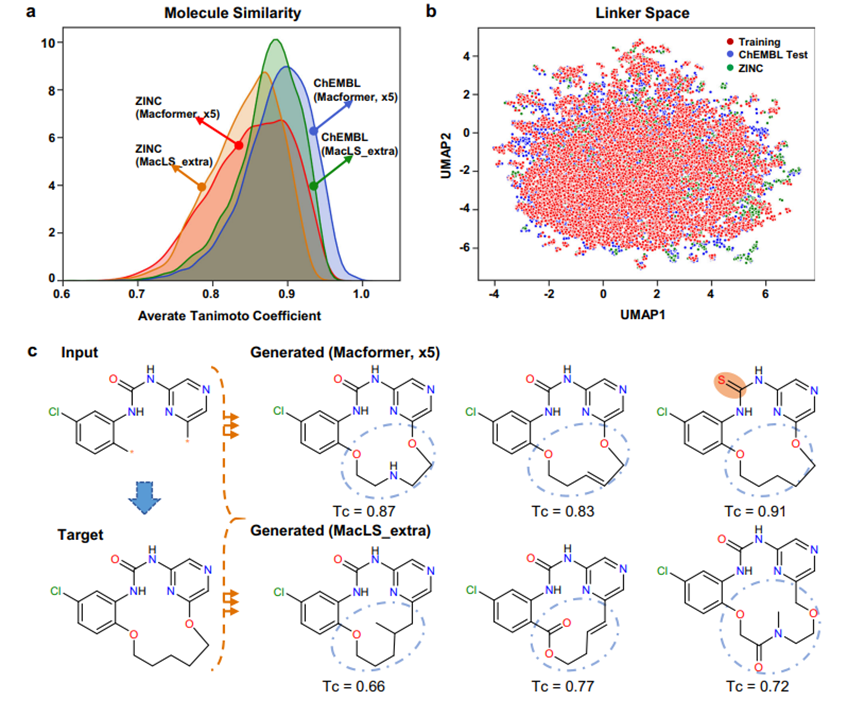
图 2. 在ChEMBL测试数据集和ZINC数据集上,分别使用五倍数据增强训练的Macformer和MacLS_extra生成的新型大环化合物之间的化学空间比较
为了揭示 Macformer 在这个特定的自动宏环化任务中的工作原理,分别从子串和标记尺度分析了输入和输出序列之间的注意力权重(图 3)。输入序列中的子串或标记往往对预测序列中相同子串或标记的生成影响最大,这保证了生成的宏循环中起始非循环片段的再现。当推断大环链接子串时,作者的模型显示出系统的方式,因为源序列的不同子串之间的注意力权重差异并不显着。这表明 Macformer 能够组合输入非循环 SMILES 序列的潜在特征,并将适当的连接子合并到原始线性片段中。这种能力源于它已经了解的关于训练数据集中的非循环片段与其相应的大环连接子之间的关系的先验知识。
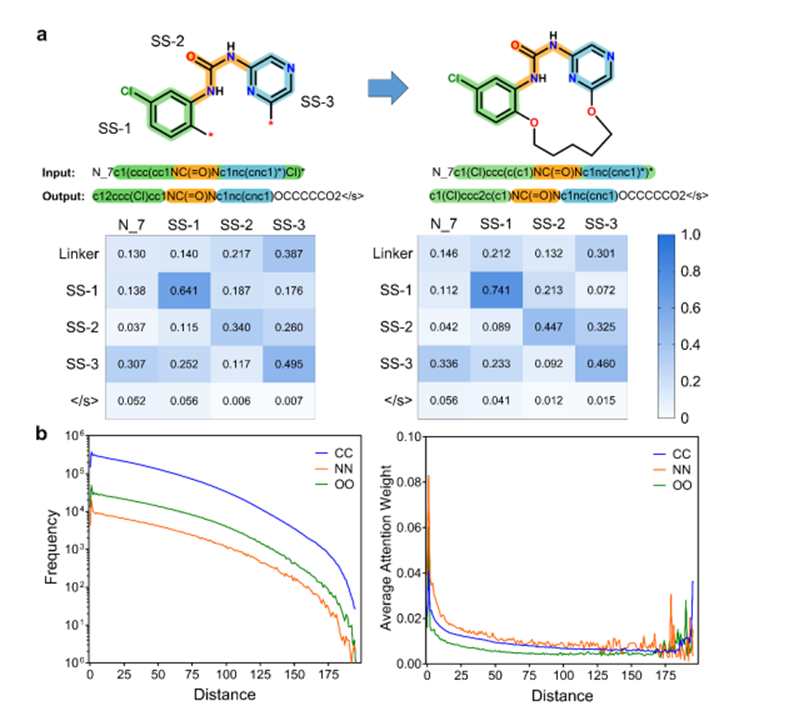
图3.注意力权重分析
JJAK2属于细胞内非受体蛋白酪氨酸激酶JAK家族激酶,是治疗骨髓增生性肿瘤和类风湿性关节炎的重要靶点。为了进行前瞻性评估,我们使用Macformer设计了大环化合物JAK2抑制剂--Fedratinib,为线性小分子化合物。但Fedratinib在整个激酶谱中的选择性并不尽如人意,在两项分别以结合亲和力KD和半数抑制浓度IC50为指标的系统性研究中,其对多个激酶均有活性。为改善Fedratinib的激酶选择性,采用Macformer对Fedratinib进行环化,最终获得218个新型大环分子,结合分子对接和基于经验的合成可行性分析,选择3个具有不同骨架的大环化合物进行化学合成和生物活性验证(图4)。其中,活性最好的化合物3对JAK2的激酶水平抑制活性在单个纳摩尔级别(IC50 = 0.006±0.001 μM),与Fedratinib活性相当(IC50 = 0.003±0.001 μM)。在细胞抗增殖测试中,新合成的大环化合物也表现出较好的活性。
在采用MacLS对Fedratinib进行环化获得的300个化合物中,未发现经Macformer大环化得到的3个活性较好的JAK2抑制剂。实际上,Macformer和MacLS生成的化合物之间的交集只有2个大环化合物,表明Macformer方法可以获得被传统片段库搜索方法遗漏的活性较好的环链片段。
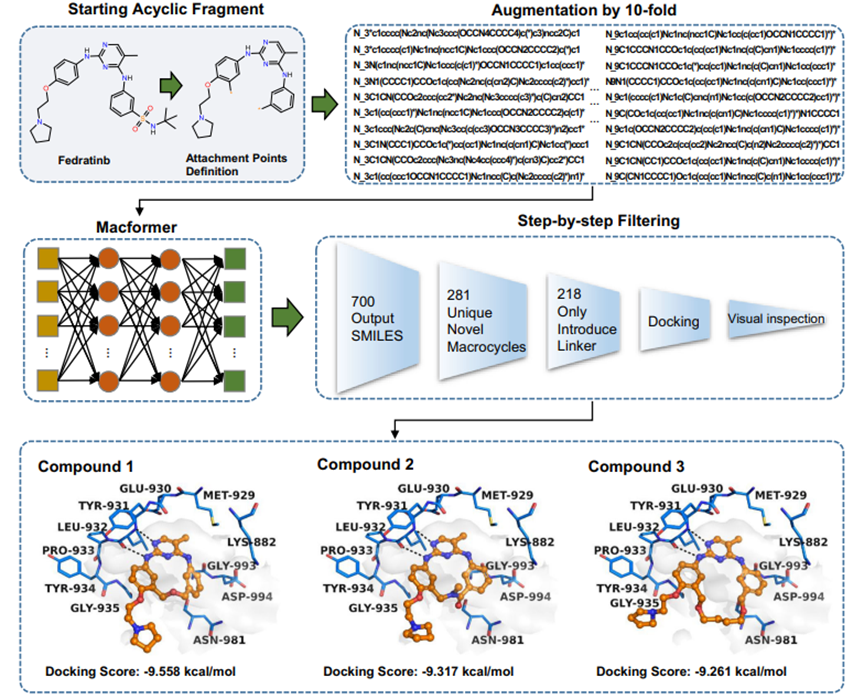
图 4.从Fedratinib开始设计大环JAK2抑制剂的流程示意图
大环化合物作为一类具有特殊骨架的分子,在药物研发领域具有巨大的应用潜力。为了利用深度学习的优势来解决大环候选药物设计问题,本研究开发了Macformer方法。Macformer能够捕获非环结构的SMILES字符串和目标大环化合物的SMILES字符串之间的隐式对应关系,并自动补齐缺失的环链,以端到端的方式自动生成大环化合物。Macformer较好的性能和泛化能力意味着其在线性分子环化方面的巨大潜力,从而为大环候选药物的设计提供了新型实用的工具,也进一步拓宽了深度学习方法在药物设计领域的应用。此外,通过对已上市JAK2激酶抑制剂Fdedratinib进行环化的前瞻性研究,验证了Macformer的实用性,也为靶向JAK2药物的后续开发提供了新颖的大环骨架。
Diao, Y., Liu, D., Ge, H. et al. Macrocyclization of linear molecules by deep learning to facilitate macrocyclic drug candidates discovery. Nat Commun 14, 4552 (2023). https://doi.org/10.1038/s41467-023-40219-8










