
本工作报道了基于人工智能的原位实验红外光谱分析,揭示合成单原子催化剂的过程机理。AI模拟的红外光谱与原实验结果相似度高达0.7-0.9,且可通过AI给出的结构相关性系数,直接从实验谱图反推化学结构,所得的反应机理与多种实验表征手段的综合结果高度一致。人工智能技术,理论计算模拟,原位实验表征的紧密结合将成为化学反应机理研究的新方法,并有望扩展到更广泛的应用。

第一作者:赵彦璋,李欢
通讯作者:李昊博
通讯作者和单位:阿德莱德大学
原文链接:https://pubs.acs.org/doi/10.1021/acs.jpclett.3c02896
关键词:原位红外光谱分析、单原子催化剂形成、ZIF 热解机理、LASSO 回归、DFT 计算数据库
人工智能技术在实验光谱分析中的有效应用,有望成为识别化学结构和反应机理的重要工具。单原子催化剂(SACs)在能源领域有着广泛的应用潜力,但对其形成机理的研究仍相对有限。此前的实验研究通过钴基沸石咪唑醇骨架(ZIF-67)的热解,成功合成了Pt掺杂的氧化钴SAC(Pt–Co3O4)。通过原位温度依赖漫反射傅立叶变换红外光谱(DRIFTS)和其他实验手段,揭示了金属-氧键的形成对SAC合成的关键作用。这些实验数据目前仍依赖于人工分析。本工作报道了一种基于机器学习的IR光谱分析技术,将最小绝对值收缩和选择算子(LASSO)回归算法应用于DRIFTS数据集,解析并对比ZIF-67和Pt掺杂ZIF-67的热解机理。通过机器学习算法与理论计算的整合,为分析原位实验光谱提供了新思路。
首先,我们构建了一个数据集,涵盖了 ZIF-67 热解过程中形成的所有可能的化学键,包括 C-C、C-N、C-O、Co(Pt)-O、Co(Pt)-N、Co(Pt)-C、Co(Pt)-Co、C-H 和 O-H 键。我们建立了不同的模型,用以反映化学键可能存在于的不同的结构环境,包括三维ZIF 模型和二维石墨烯模型等。
我们以 DFT 数据为输入,以实验数据为目标输出,通过拟合 LASSO 模型来训练机器学习模型。每个化学键的红外光谱都被视为机器学习特征(feature)。这一过程包括温度从 20 摄氏度逐渐升高到 300 摄氏度,然后在 300 摄氏度下保持 90 分钟。

Figure 1. Workflow for the data-driven IR spectroscopic analysis to extract the structural evolution mechanism using ML algorithms. The experimental IR spectrum is reproduced with permission.
训练AI的数据库包含了这些模型中的所有非等价化学键,包括 ZIF-67热解过程的55 种化学键和掺铂 ZIF-67 的86 种化学键。我们通过 DFT 计算模拟了每个化学键的红外光谱。每个化学键的数据包括 6638 个波长-吸光率对。数据库总共包含 935958 个数据点。

Figure 2. Computational models of IR spectrum database for chemical bonds during ZIF-67 (in pink frame) and Pt-doped ZIF-67 (in green frame) pyrolysis. Atoms: Co: pink-color; Pt: green; C: grey; N: blue; O: red; H: white.
AI预测的光谱与实验光谱非常相似,如图 3所示,ZIF-67 和掺铂 ZIF-67的相关值分别高达 0.7和0.9。这表明机器学习训练结果的可靠性。图4显示,我们的机器学习模型所采用的绝大多数不同特征都显示出较低的相关性。这突出说明了采用这些计算出的红外光谱作为机器学习特征的合理性。

Figure 3. ML simulated in-situ temperature-dependent DRIFTs spectra in comparison with experimental characterization outcomes for the pyrolysis processes of ZIF-67 (a-c) and Pt-doped ZIF-67 (d-e), respectively. The experimental results (a, d) exhibited a striking resemblance to the ML simulations (b, e), with correlations (c, f) reaching approximately 0.7 and 0.9, respectively.
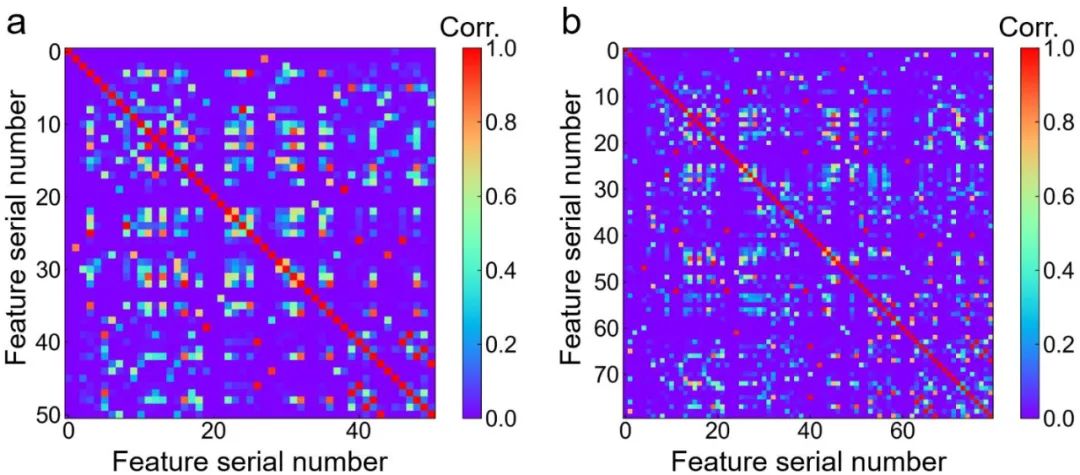
Figure 4. Pearson correlation between every pair of primary features, i.e., calculated IR spectrum for each chemical bond in ZIF-67 (a) and Pt-doped ZIF-67 (b) pyrolysis process.
图5b比较掺Pt和不掺Pt的情况。整个热解过程主要是氧化过程,其中Co和Pt原子最初与ZIF框架内的四个甲基咪唑配体的N原子形成连接。随着ZIF骨架的分解,形成了平面的CoN4或PtN4结构。在高温300°C下持续一段时间后,四个N原子逐渐脱离,与此同时形成Co-O键(图5a中的绿线)。相比之下,Pt-O键的系数明显比Co-O键增加得更快(图5b中的粉红线),表明Pt-O键的形成比Co-O键更早。这与实验观察一致,即从Co-N到Co-O的转变大约在300°C左右开始,之前Pt-N逐渐转变为Pt-O。然后,这些Pt-O和Co-O键相互连接,最终形成含有高密度Pt单原子的Pt–Co3O4催化剂结构。

Figure 5. Coefficients of selected chemical bonds along the temperature-time axis for the pyrolysis processes of ZIF-67 (a) and Pt-doped ZIF-67 (b). The model structures corresponding to the different colors of the chemical bonds are labeled below, and they are positioned along the temperature-time axis based on the approximate peak positions of the coefficients. It illustrates the evolution process of the structures. Atoms: Co: pink-color; Pt: green; C: grey; N: blue; O: red.
理论计算的光谱虽然与实验测得的光谱之间有一定差异,但鉴于二者在原理上的相关性,可以利用机器学习方法弥补他们之间的差异。通过理论计算数据库训练的AI模型,能够给出在模拟实验光谱时所选用不同结构以及其相关性系数。如果一种结构的相关性系数下降,同时另一种结构的相关性系数上升,那么很有可能是前一种结构转化成了后一种结构。通过这种方法观察到的 ZIF 框架降解、吸附在金属上的含氧物种逐渐氧化以及 Co-O 和 Pt-O 键的出现与各种实验表征的结果非常吻合。值得强调的是,我们开发的整个工作流程在很大程度上依赖于数据库构建和机器学习算法,人工干预极少。因此,我们相信这种方法具有很好的稳健性,未来有巨大的潜力扩展到广泛的应用领域,对原位实验表征数据进行智能分析。

李昊博博士:阿德莱德大学化工学院特聘研究员、博士生导师。2012和2017年分别在南开大学化学学院和中国科学院大连化学物理研究所获得理学学士和博士学位。2018-2021年获德国洪堡基金资助,在慕尼黑工业大学、德国马普协会Fritz-Haber研究所进行合作研究。2023年入选《麻省理工科技评论》“35岁以下科技创新35人”(MIT TR35)亚太区。2024年获澳大利亚研究理事会优秀青年基金(ARC DECRA)资助。在Science, Nat. Commun., J. Am. Chem. Soc., Angew. Chem. Int. Ed., Adv. Mater.等期刊上发表论文50余篇,被引用7700余次。主要从事理论催化研究,方向包括能源催化转化的从头计算模拟、人工智能驱动能源材料设计等。
相关文章
(1) Li, H.; Jiao, Y.; Davey, K.; Qiao, S. Z. Data‐driven machine learning for understanding surface structures of heterogeneous catalysts. Angew Chem. Int. Ed. 2023, 135, e202216383.
(2) Mairegger, T.; Li, H.; Grießer, C.; Winkler, D.; Filser, J.; Hörmann, N. G.; Reuter, K.; Kunze-Liebhäuser, J. Electroreduction of CO2 in a non-aqueous electrolyte-the generic role of acetonitrile. ACS Catal. 2023, 13, 5780-5786.
(3) Shan, J.; Liao, J.; Ye, C.; Dong, J.; Zheng, Y.; Qiao, S. Z. The dynamic formation from metal-organic frameworks of high-density platinum single-atom catalysts with metal-metal interactions. Angew. Chem. Int. Ed. 2022, 61, e202213412.
文本编辑:最后的道哥哥

科学温故QQ群—科研爱好者集中地!(不定期发布讲座通知,分享录制视频)微信群(学术交流/电催化/光催化/理论计算/资源共享/文献互助群;C1化学/生物质/单原子/多孔材料分舵),小编微信:hao-xinghua或alicezhaovip,备注“姓名-单位”










