将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

编辑 | 萝卜皮
高度多重蛋白质成像正在成为分析细胞和组织内天然环境中蛋白质分布的有效技术。然而,现有的利用高复杂空间蛋白质组学数据的细胞注释方法是资源密集型的,并且需要迭代的专家输入,从而限制了它们对于广泛数据集的可扩展性和实用性。
哈佛医学院(Harvard Medical School)团队引入了 MAPS(Machine learning for Analysis of Proteomics in Spatial biology),这是一种机器学习方法,有助于从空间蛋白质组数据中快速、精确地识别细胞类型,并具有人类水平的准确性。
MAPS 在多个内部和公开可用的 MIBI 和 CODEX 数据集上进行了验证,在速度和准确性方面优于当前的注释技术,即使对于通常具有挑战性的细胞类型(包括免疫来源的肿瘤细胞)也能达到病理学家级别的精度。
该研究以「MAPS: pathologist-level cell type annotation from tissue images through machine learning」为题,于 1 月 2 日发布在《Nature Communications》。

细胞亚型的精确描述对于阐明生物组织在其自然环境中的结构和功能的复杂性至关重要。高重空间蛋白质组学技术(例如 MIBI、CODEX、cycIF 和 IMC)的最新进展,允许在单个组织切片内询问 40-60 个蛋白质组标记,为表型和功能研究的细胞和组织结构内的蛋白质表达和分布提供了宝贵的见解。
然而,这些方法可能面临与组织降解、图像配准困难以及循环过程中表位丢失相关的障碍。
高度多重的图像可以为了解生物过程提供新的方向,但它们也给数据处理带来了挑战,比如需要自动化管线从每个单细胞中提取信息。现有的细胞注释方法取决于无监督的聚类技术,需要随后的手动管理和视觉验证,这个过程可能明显是劳动密集型的,并且需要特定领域的专业知识。
因此,需要一种计算量轻、快速的自动化细胞分类方法,在达到人类水平的准确性的同时,提高空间蛋白质组数据分析的效率和可扩展性。
哈佛医学院的研究团队开发了 MAPS(Machine learning for Analysis of Proteomics in Spatial biology),这是一种机器学习包,可在跨多个空间蛋白质组学平台进行基准测试时实现准确、快速的细胞注释,并具有最高的同类性能。MAPS 可以提高细胞注释过程的速度和质量,以便研究人员可以分配更多的下游工作来原位揭示新的生物过程。
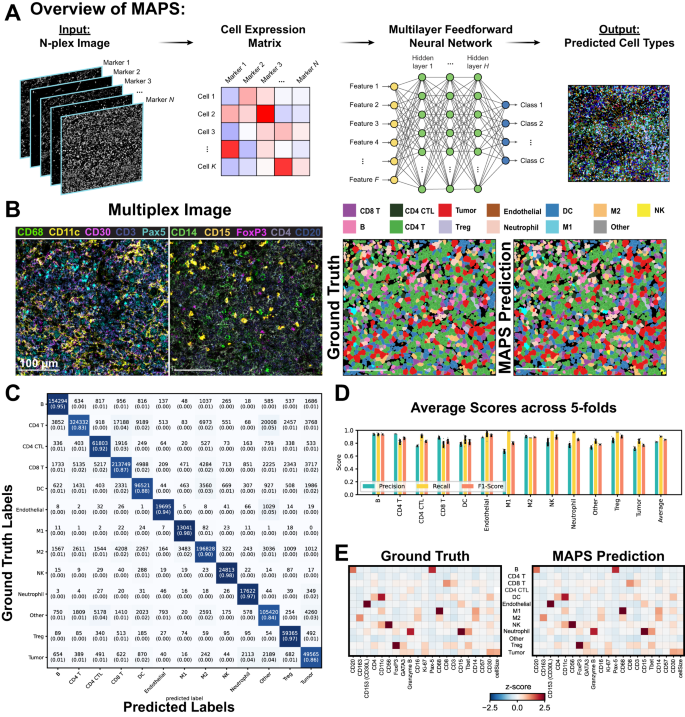
图示:MAPS 架构及其在 cHL1 (MIBI) 数据集上跨 5 倍交叉验证的性能概述。(来源:论文)综合评估表明,MAPS 在准确性和计算效率方面均优于其同类产品 ASTIR 和 CellSighter,从而使其成为精确细胞类型预测的强大工具。
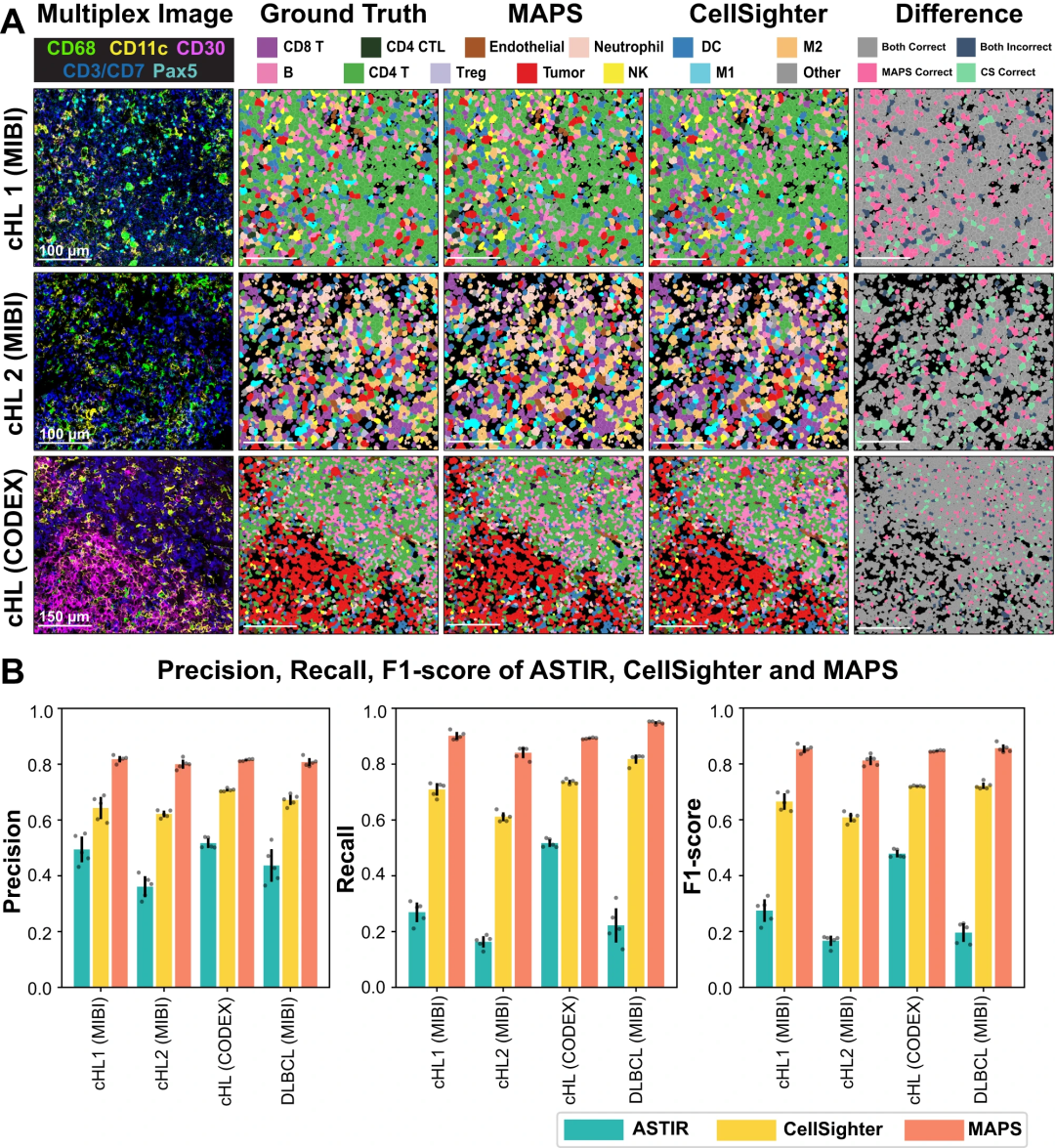
图示:MAPS 性能与同类产品比较。(来源:论文)
与现有最先进的方法相比,MAPS 表现出卓越的性能指标。具体来说,它实现了显著更高的 F1 分数、精确度和召回率,展示了其从空间蛋白质组数据中准确辨别细胞类型的卓越能力。这种提高的性能证明了 MAPS 中采用的前馈神经网络架构的有效性。
这种架构能够有效处理空间蛋白质组数据,从而捕获输入特征和细胞类型之间的复杂关系。ReLU 激活函数的结合引入了非线性,进一步增强了模型识别复杂细胞模式的能力。训练过程中 dropout 层的集成可以减轻过度拟合,增强模型的泛化能力。
MAPS 的优势在于其在不同的生物环境中始终保持高性能。它展示了处理各种疾病模型的熟练程度,例如经典霍奇金淋巴瘤 (cHL)、弥漫性大 B 细胞淋巴瘤 (DLBCL) 和结直肠癌 (CRC)。这种适应性展现了 MAPS 的多功能性,使其成为广泛的生物和生物医学研究应用的可靠工具。

图示:定量比较 MAPS 应用于外部数据集和跨数据集时的性能。(来源:论文)
此外,MAPS 表现出卓越的跨平台兼容性,在 MIBI 和 CODEX 数据集上始终表现良好。这一功能至关重要,因为它确保了 MAPS 在不同实验环境中的适用性。跨数据集的合理水平的通用性,进一步巩固了 MAPS 作为空间蛋白质组数据细胞注释的领先方法的地位。
在数据效率方面,除了在充分采样的场景中表现出色之外,MAPS 在使用有限的训练数据进行训练时也表现出一致的性能。即使在数据可用性可能受到限制的情况下,此功能也可以实现准确的细胞类型注释。只要注释的单元格能够很好地代表其各自的群体,使用中等大小的数据集就可以实现 MAPS 的最佳性能。
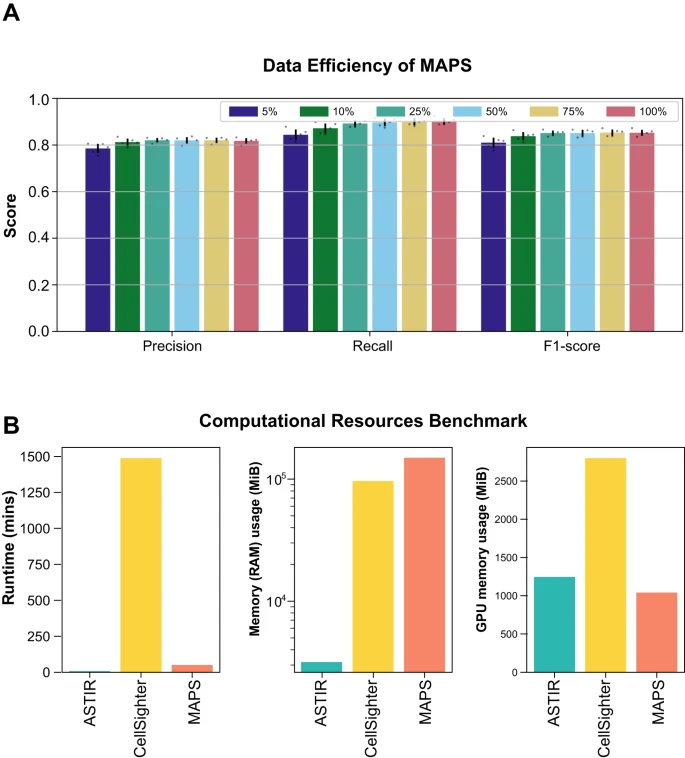
图示:MAPS的数据效率和计算效率。(来源:论文)
最后,MAPS 不仅在准确性方面超越了同类产品,而且在计算效率方面也脱颖而出。其训练时间比现有的监督方法快几个数量级,这是分析大规模空间蛋白质组数据的关键优势。
这种效率是一个关键特征,特别是在必须快速处理大量数据集的情况下。通过将 MAPS 集成到当前的空间蛋白质组学工作流程中,它可以加快较小的、精心策划的「地面实况」数据集的注释过程,证明了其简化该领域研究工作的潜力。
总之,卓越的性能、简单的模型架构、快速训练和推理、跨平台兼容性以及对不同组织类型和疾病模型的适应性相结合,使 MAPS 成为空间蛋白质组数据细胞注释的强大工具。
MAPS 包和 GitHub 上相关数据资源的发布标志着对科学界的重大贡献,为研究人员提供了宝贵的资源,以推进组织空间组学领域的发展,并加速跨不同生物背景的细胞生物学的发现。
开源地址:https://github.com/mahmoodlab/MAPS
论文链接:https://www.nature.com/articles/s41467-023-44188-w
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。









