最近有小伙伴反映收不到推送,因为公众号改了推送算法,现在需要加星标,多点赞、点在看,才能准时收到推送哦。
导语:图表示学习已被用于从生物网络中识别癌症基因。然而,它的适用性受到综合网络分析下可解释性和可推广性不足的限制。
作图丫不仅文章解读的好,课题做得也出色,已与国内多家知名医院的老师和名牌大学实验室达成合作。欢迎有生信分析需求的老师垂询,公共数据库数据挖掘或自测数据分析均可。

今天小编为大家带来的这篇文章,报告了一种可解释和可推广的基于 Transformer 的模型的开发,该模型通过利用图表示学习以及将多组学数据与同质和异质生物相互作用网络的拓扑结构相结合,准确预测癌症基因。文章发表在《nature biomedical engineering》,文章题目为:Interpretable identification of cancer genes across biological networks via transformer-powered graph representation learning。
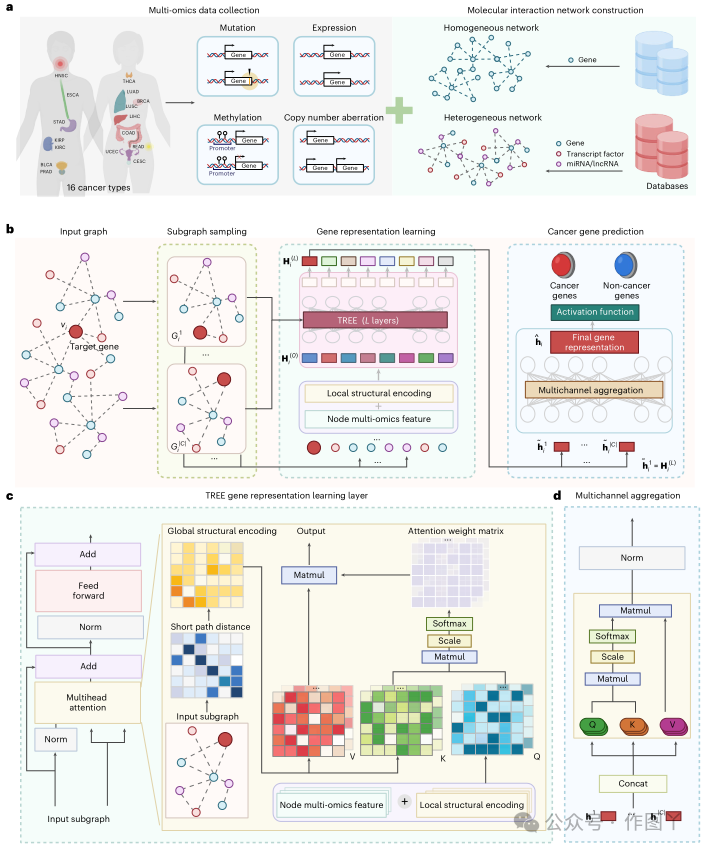
图 1
a、多组学数据收集和同质/异构网络构建。对于基因和转录因子,共收集了 16 种癌症类型的 4 种组学数据,包括基因突变、表达、甲基化和拷贝数变异,而对于 miRNA 和 lncRNA,仅收集了表达数据。此外,还收集并构建了两类网络,包括由基因组成的同质网络,以及由基因、转录因子和 lncRNA/miRNA 组成的异构网络。b、癌症基因预测图解。对于目标基因,TREE 首先在其局部结构内采样多子图,然后将子图输入基因表征学习模块,以在每个子图上生成基因表征。基因表征学习中的参数由所有子图共享,该图仅显示了 𝒢𝒢1 i 的学习过程。在获得每个子图上的基因表示后,创建多通道聚合模块进行融合得到最终的基因表示,通过激活函数判断目标基因是否致癌。c,TREE基因表示学习层示意图。左图为基因表示学习层的组成,输入为子图,以共同注意的方式实现多头注意力,如右图所示。通过生成query和节点多组学特征将网络局部结构信息编码到TREE中,通过生成key和value将全局结构信息集成到TREE中。d,多通道聚合模块示意图。其架构与传统Transformer相同,即以SA的方式实现多头注意力。TREE在识别各种环境下的癌症基因方面表现出普遍性为了证明 TREE 的有效性,本研究在 8 个生物泛癌症网络和 31 个癌症特定网络上进行了实验。此外,本研究在相同的实验设置下将 TREE 与 5 种基于 SOTA 网络的方法(包括拉普拉斯 (LAP)、DeepWalk (DW)、LINE、EMOGI 和 GAT)的性能进行了比较。在所有泛癌症和癌症特定网络中,TREE 的精确召回曲线下面积 (AUPRC) 值均超过 5 种比较方法(图 2a、b)。此外,TREE 在同质和异质癌症特异性网络上均表现出色,AUPRC 值平均分别提高了 7.31% 和 11.28%,如图 2b 所示。所比较方法在不确定性方面的不同表现反映了它们在推广到不同生物网络方面的局限性。在这方面,TREE 优于其他方法,因为它可以推广到所有基准网络。本研究还使用不同的颜色来区分癌症和非癌症基因,这些基因基于多组学特征的 t 分布随机邻域嵌入 (t-SNE) 表示。因此,可以反映真实结果和预测结果之间的匹配程度,以指示比较方法的分类准确性(图 2c)。结果表明,所提出的 TREE 在基因分类方面达到了最高的准确率。此外,本研究还通过考虑所有网络展示了每种比较方法的分类置信度得分(图 2d)。特别是,TREE 分别以 0.9 和 0.1 的置信度得分预测癌症和非癌症基因,而其他方法在对已知癌症基因和非癌症基因进行分类方面不如 TREE。如上所述,本研究中使用的同质和异构网络都是不平衡网络,这导致 LAP、DW 和 LINE 在非癌症基因中的表现更好。这一发现再次表明了 TREE 在癌症基因识别任务中的可靠性。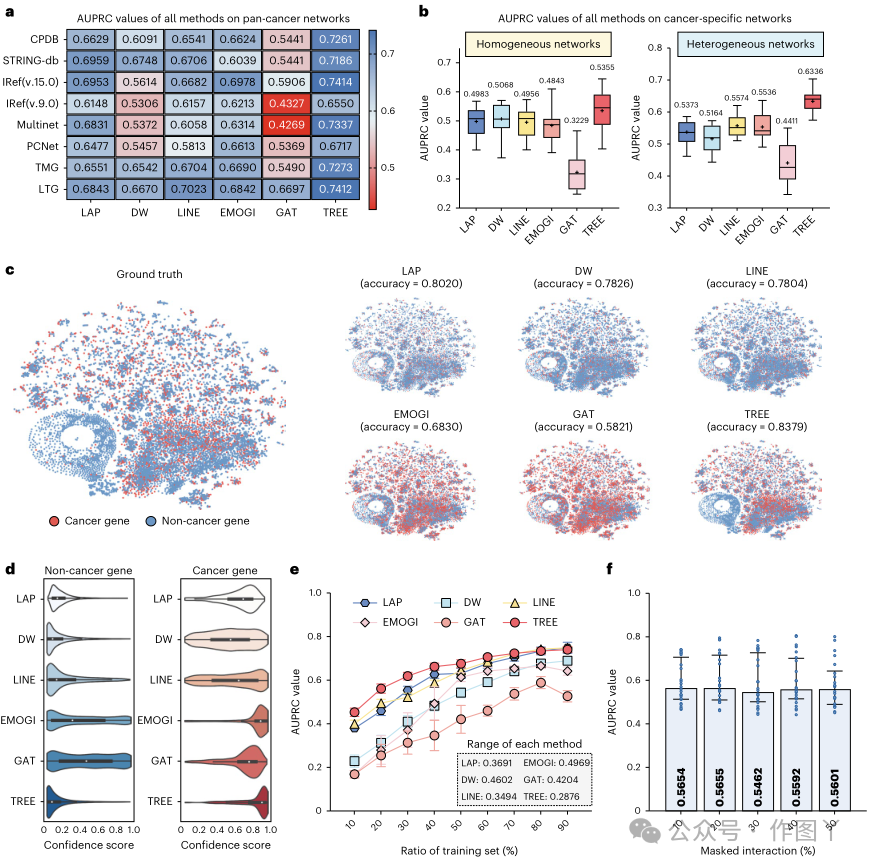
图 2
为了展示 TREE 的稳健性,本研究采用少样本学习来评估其性能,以及所有基线模型在各种数据集上的性能。具体来说,本研究随机选择 10% 到 90% 的标记基因作为训练集,而其余基因构成测试集。图 2e 中显示的结果说明了每个模型在不同训练集大小比率下的性能。TREE 始终优于所有基线方法,证明了其在各种训练集比率中均具有卓越而稳定的性能。此外,本研究通过从原始网络中随机删除 10% 到 50% 的交互来评估其稳健性。图 2f 中所示的结果显示,虽然 TREE 的性能可能会在不同设置下波动,但变化很小。为了阐明四类不同组学数据的影响,本研究使用模型梯度来评估它们的重要性,如方法部分所述。如图 3a 所示,该评估是在泛癌场景和癌症特定场景下进行的。研究结果揭示了突变在辨别泛癌基因中的关键作用。此外,在分析影响癌症基因识别的决定因素时,本研究发现,在全癌症和癌症特定情况下,所有四种类型的组学数据都有助于基因识别。图 3b 中显示了它们始终如一的高置信度得分。值得注意的是,这一发现也有助于理解将某些基因指定为癌症基因背后的原理。随后,本研究辨别出癌症基因和非癌症基因之间 SNV、基因表达 (GE) 和 CNA 的显著区别,如图 3c 所示。受此观察的启发,本研究试图研究组学数据是否可以作为识别癌症基因的信号。为了探索这一点,本研究
根据组学数据的值将整个数据集拆分为训练集和测试集,建立了四种不同的场景。这些场景称为 SNV 拆分、甲基化 (METH) 拆分、GE 拆分和 CNA 拆分。以 SNV 拆分为例,本研究根据排名组织基因突变值,制定包含前 80% 基因的训练集,并将剩余的 20% 分配给测试集。随后,本研究使用训练集在 10 倍交叉验证设置中训练模型,并评估其在测试集上的性能。结果如图 3d 所示。值得注意的是,与随机分割相比,TREE 和所有基线模型在这些新创建的场景下都表现出更好的性能,这表明组学数据确实可以被视为识别癌症基因的显著信号。这在基线模型在新场景下的性能大幅提升中得到了证明,因为它们倾向于识别具有高突变、表达水平、拷贝数改变或甲基化的基因。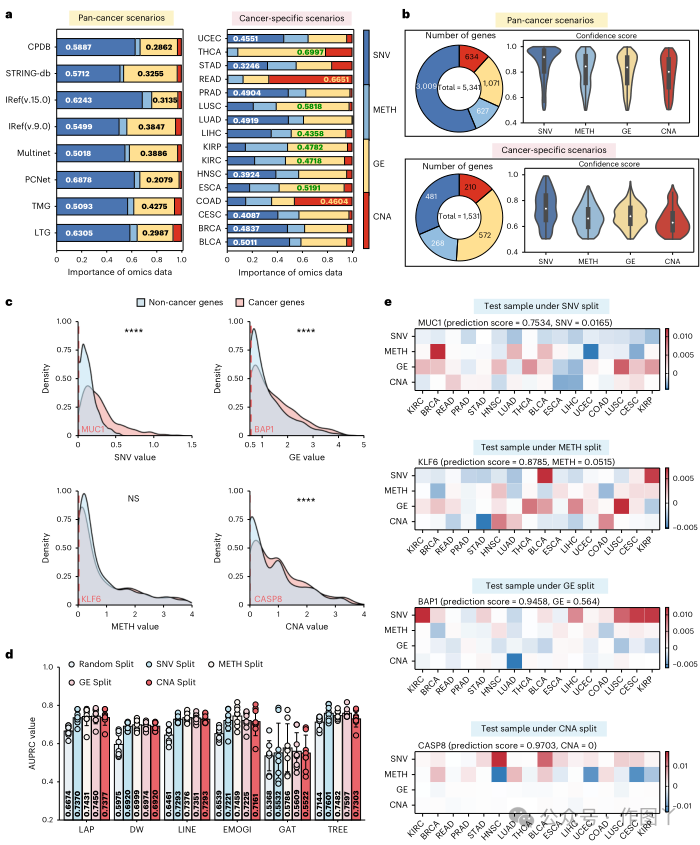
图 3
然而,与基线模型相比,本研究提出的 TREE 模型表现出卓越的稳健性,成功识别了具有低组学值的癌症基因,如图 3d 所示。这种增强的稳健性得到了进一步强调,因为 TREE 的表现始终优于所有基线模型,在不同场景中保持稳定性。值得注意的是,TREE 展示了其在识别具有低组学值的癌症基因方面的能力,如图 3e 所示。为了说明这一点,本研究在不同场景下选择了四个基因(MUC1、KLF6、BAP1 和 CASP8),它们作为测试样本,并且未被本研究中检查的任何其他基线模型检测到。尽管它们在相应的组学分割场景中的值很低,但 TREE 成功地通过其他组学数据识别了这些基因,如图 3e 所示的组学数据重要性所证明。这一发现不仅证实了 TREE 的有效性,还揭示了将这些基因归类为癌症基因的原因。为了进一步研究不同基因相关调控机制的影响,本研究定义了总共 7 个简单和复杂的元路径,模拟不同类型分子相互作用之间的相互依赖性。本研究使用每个元路径来指示调节癌症基因的可能功能机制。如图 4a 所示,然后构建一个子网络,并从原始异质网络中提取其所有实例。本研究通过评估 TREE 在其相应子网络上的性能来研究元路径的影响。GG 元路径的实例、GG 网络与同质配置对齐。然后,本研究通过从使用这些定义的元路径生成的网络中学习基因嵌入来评估 TREE 识别癌症基因的性能。为了直观地比较 TREE 与使用不同元路径生成的子图的性能,本研究在箱线图中展示了它们的 AUPRC 值(图 4b、c)。总体而言,所有元路径的整合使 TREE 在 TMG 和 LTG 上的性能最佳,AUPRC 值分别高出 14.31% 和 4.345%,这表明依赖单个调控机制可能不足以从整体角度识别所有癌症基因。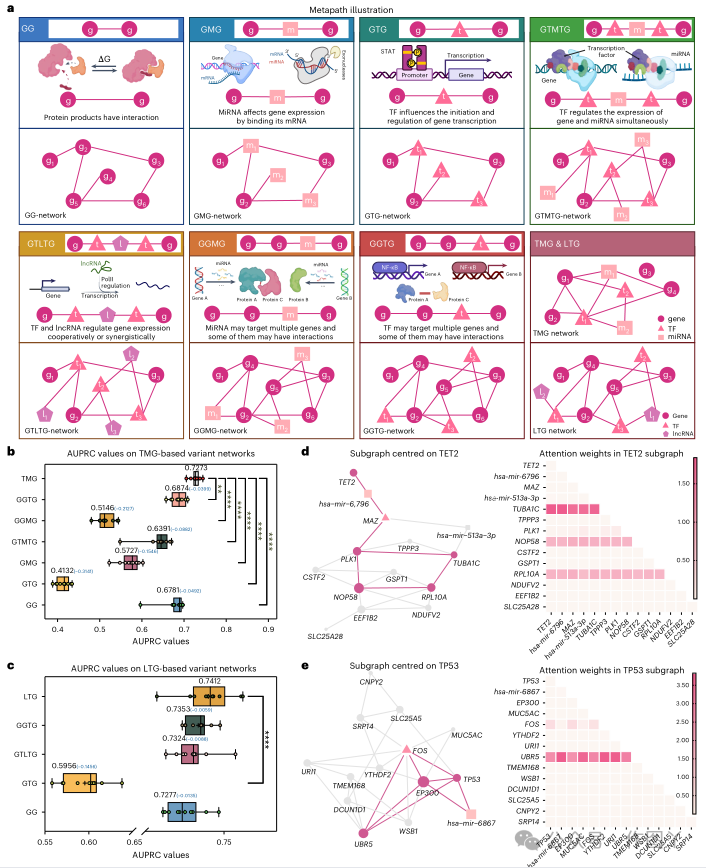
图 4
本研究还提供了两个案例研究来进一步说明 TREE 如何受益于各种调控机制,如图 4d、e 所示。只有通过从所有元路径组合中学习到的基因嵌入才能正确预测 TET2 基因。TREE 通过遵循复杂的调控机制成功识别了它,网络路径为基因–miRNA–TF–基因–基因。图 4d 中提供的调控途径表明 hsa-mir-6796 靶向 TET2 和 MAZ,调节 MAZ 相关基因(PLK1 和 TUBA1C)以及 TET2 的表达,从而影响 MAZ 控制的细胞周期调控和 TET2 肿瘤抑制功能中的细胞过程。此外,本研究还以另一个著名的泛癌基因 TP53 为例,进一步表明所识别的网络模式的有效性。如图 4e 所示,UBR5 在 TP53 的识别中起着重要作用。 最短路径信息赋予了自适应优化基因感受野图结构的能力接下来,本研究将重点根据从 TREE 获得的注意力权重来指定用于癌症基因识别的最重要图形模式。具体来说,对于每个正确预测的基因,本研究首先从 TREE 中提取其所有注意力权重矩阵,然后计算其在每层头上的平均注意力权重矩阵。在研究结果后,本研究发现 TREE 能够通过捕获不相交节点的长依赖关系来自适应地学习基因嵌入。以基因 TP53 为例,它被正确预测为具有最高置信度得分的癌症基因。如图 5a 所示,本研究将其感受野的图形结构可视化,以逐层说明决策过程。从图 5a 中可以明确地注意到,考虑最短路径信息赋予了即使在不相交的节点之间也能对长依赖关系进行建模的能力。更重要的是,这些长依赖关系对于基因嵌入学习至关重要。例如,在图 5a 中显示的最后一层中,尽管基因 CTBP2、KRT18、TLE2 和 PDCD10 与 TP53 没有直接联系,但它们对正确预测 TP53 的贡献最大。因此,从全局角度识别癌症基因时可以验证长依赖关系的重要性和必要性。由于实验已经证实了 TREE 在同质和异构生物网络上的普遍性,本研究打算研究这两类网络是否具有相同或相似的图形模式以正确识别癌症基因。为此,本研究首先在相同的实验设置下在每个基准网络上训练 TREE。然后,本研究通过记录最短路径的长度以及每层中成对节点之间的注意权重,分析所有正确识别的癌症基因在六个同质和两个异构网络中的决策过程。最后,本研究根据节点对的网络距离对其进行分组,并在图 5b 中展示每层中所有组的平均注意权重。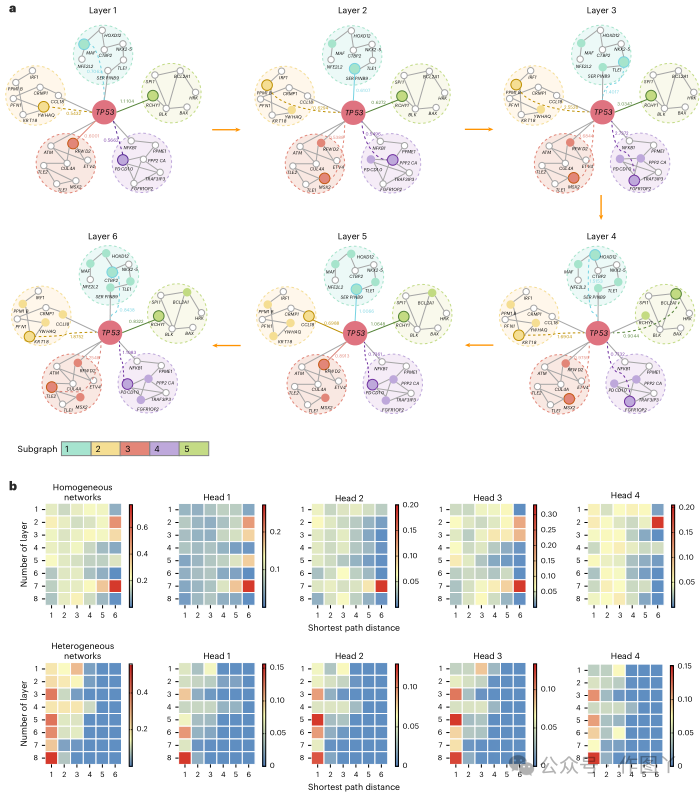
图 5
为了发现新的癌症基因候选物,本研究部署了 TREE 来评估 4,729 个未标记基因,这些基因存在于所有 8 个网络中。本研究的预测分析显示,其中 57 个基因在所有数据集中的最低置信度得分超过 0.9。这些发现促使本研究推荐它们作为本研究中的潜在癌症基因候选物,如图 6a 所示。为了评估本研究预测的可靠性和可重复性,本研究对 TREE 在所有数据集中为 4,729 个常见未标记基因和 57 个潜在癌症基因候选物获得的预测分数进行了统计分析。此外,本研究计算了这些基因在多个数据集中的相关性。预测分数和相关性分数的分布如图 6b、c 所示。值得注意的是,本研究的研究结果表明,TREE 在两种情况下在所有数据集中始终表现出很强的相关性(4,729 个常见基因的相关性为 0.43–0.65,57 个推荐的癌症基因候选基因的相关性为 0.20–0.65)。这种一致性凸显了 TREE 的稳定性和可靠性,使其成为实际应用的可行模型。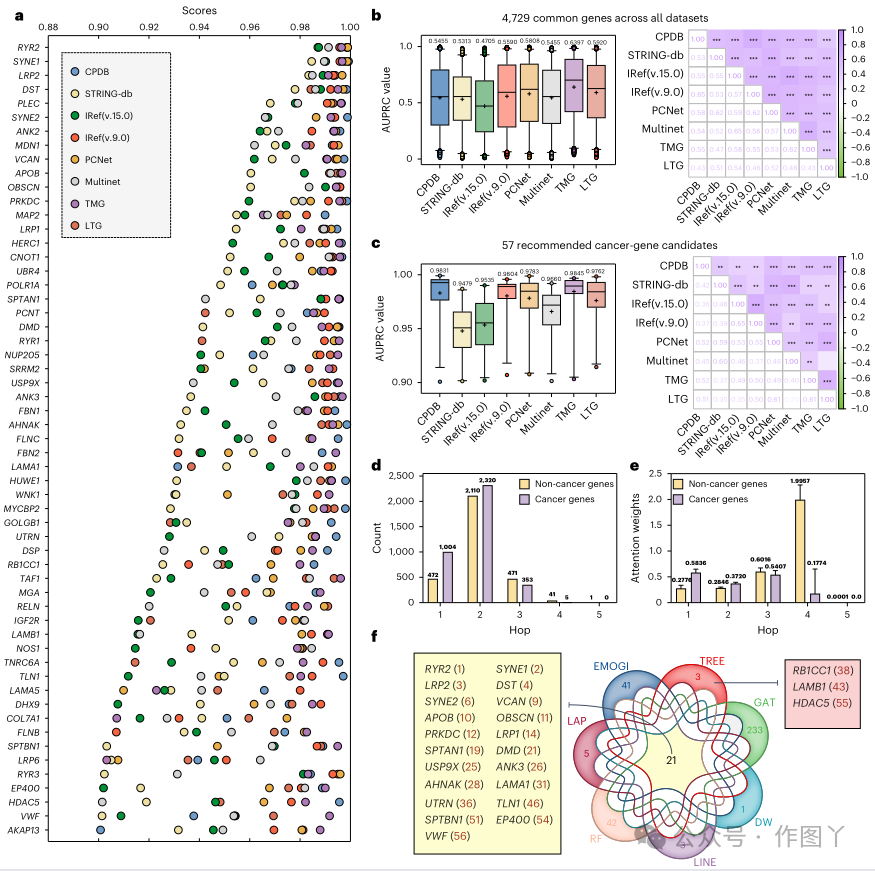
图 6
该模型可以解释多组学和高阶结构特征的各自重要性,在泛癌症和癌症特定场景中跨生物网络(包括 miRNA 与蛋白质、转录因子与蛋白质以及转录因子与 miRNA 之间的相互作用网络)预测癌症基因方面取得了最先进的性能,并在 8 个泛癌症数据集中的 4,729 个未标记基因中预测了 57 个癌症基因候选基因(包括三个未被其他模型识别的基因)。该模型的可解释性和泛化性可能有助于理解基因相关的调控机制和发现新的癌症基因。
码字不易,欢迎读者分享或转发到朋友圈,任何公众号或其他媒体未经许可不得私自转载或抄袭。由于微信平台算法改版,公众号内容将不再以时间排序展示,建议设置“作图丫”公众号为星标,防止丢失。星标具体步骤为:(2)点击右上角的小点点,在弹出界面选择“设为星标”即可。










