将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑 | 萝卜皮
基于质谱的蛋白质组学中肽段鉴定对于理解蛋白质功能和动力学至关重要。传统的数据库搜索方法虽然应用广泛,但依赖于启发式评分函数,必须引入统计估计才能获得更高的鉴定率。
加拿大滑铁卢大学 (University of Waterloo)和中原人工智能研究院(中原 AI 院)的研究团队提出了 DeepSearch,一种基于深度学习的串联质谱端到端数据库搜索方法。DeepSearch 利用对比学习框架下改进的基于 Transformer 的编码器-解码器架构。
与依赖离子间匹配的传统方法不同,DeepSearch 采用数据驱动的方法来对肽谱匹配进行评分。DeepSearch 还可以以零样本方式分析可变的翻译后修饰。
团队在各种数据集中验证了 DeepSearch 的准确性和稳健性,包括来自蛋白质组成多样的物种的数据集和富含修饰的数据集。这为串联质谱中的数据库搜索方法提供了新的启示。
该研究以「Towards highly sensitive deep learning-based end-to-end database search for tandem mass spectrometry」为题,于 2025 年 1 月 6 日发布在《Nature Machine Intelligence》。

基于质谱(MS)的蛋白质组学中,肽鉴定是一项基本挑战,通常通过将实验获得的 MS/MS 光谱与理论光谱进行数据库搜索匹配。然而,现有方法依赖启发式评分函数,可能忽略大量碎片信息,需引入概率模型提高鉴定率。
近年来,深度学习技术如 DeepNovo 和 PointNovo 显著提升了从头肽测序的准确性,但仍面临蛋白质组成差异大和翻译后修饰识别不足的挑战。
最近引入的对比学习框架下的多模态基础模型显著提高了各种下游跨模态理解任务的性能,尤其是在计算机视觉和自然语言处理领域。这些模型能够学习跨不同模态的联合嵌入空间,并在零样本学习任务中表现出色。
最重要的是,这些框架下的弱监督机制不需要跨模态数据对以外的注释,从而提高了对偏差的容忍度和增强了跨数据集的稳健性。
在最新的研究中,研究人员提出了第一个基于深度学习的端到端数据库搜索方法 DeepSearch。DeepSearch 采用跨模态余弦相似度作为评分方案,而不是离子到离子匹配。
DeepSearch 在对比学习框架下进行训练,并与 MassIVE v2 上的从头测序目标联合优化,MassIVE v2 是一组基于人类 MS/MS 库构建的高质量肽谱匹配(PSM)。
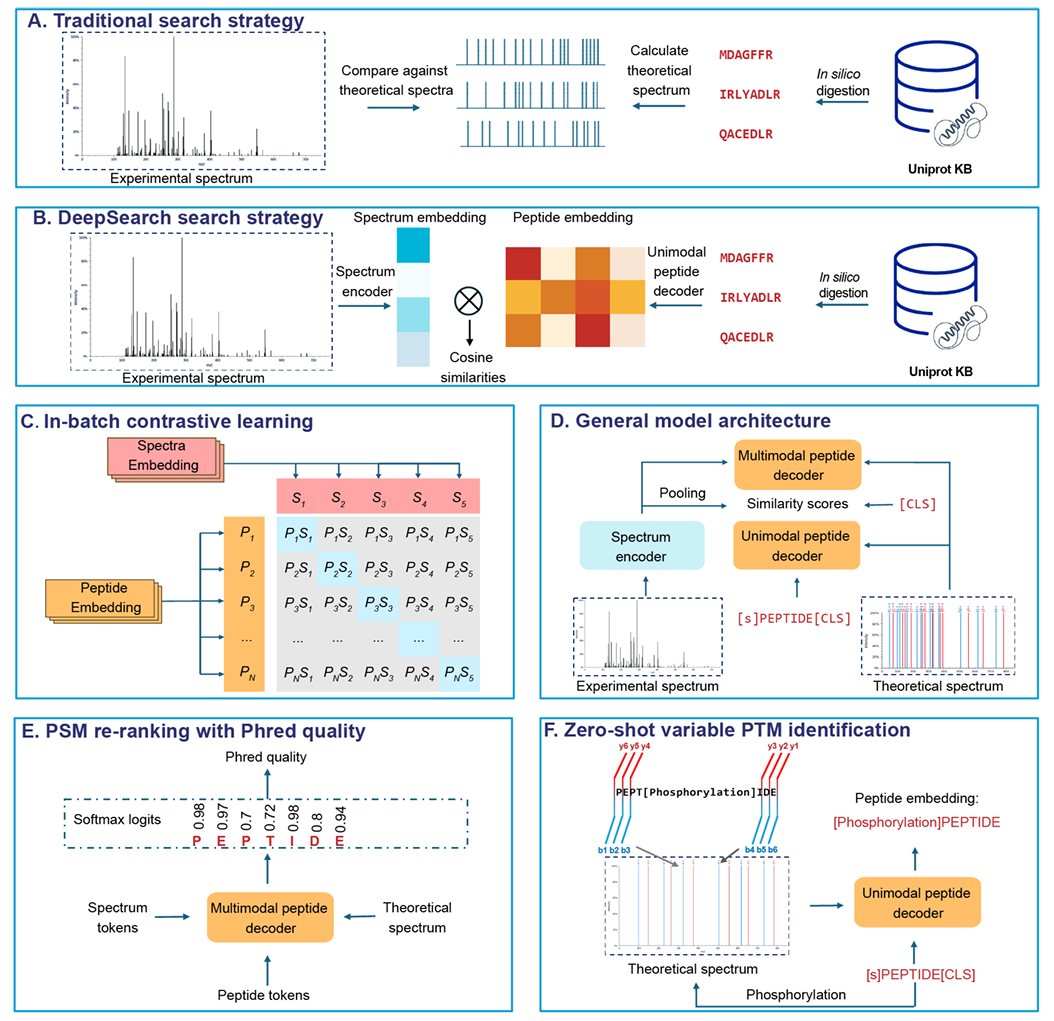
图示:数据库搜索策略和 DeepSearch 模型。(来源:论文)
为了解决在训练数据中注释 PSM 的负对和与搜索引擎算法相关的偏差的挑战,DeepSearch 采用了批量对比学习框架,该框架具有质量锚定采样方案。
与执行离子对离子匹配的传统数据库搜索引擎不同,DeepSearch 使用光谱和肽嵌入之间的余弦相似性对 PSM 进行排序,从而可以通过单个矩阵乘法进行高效计算。
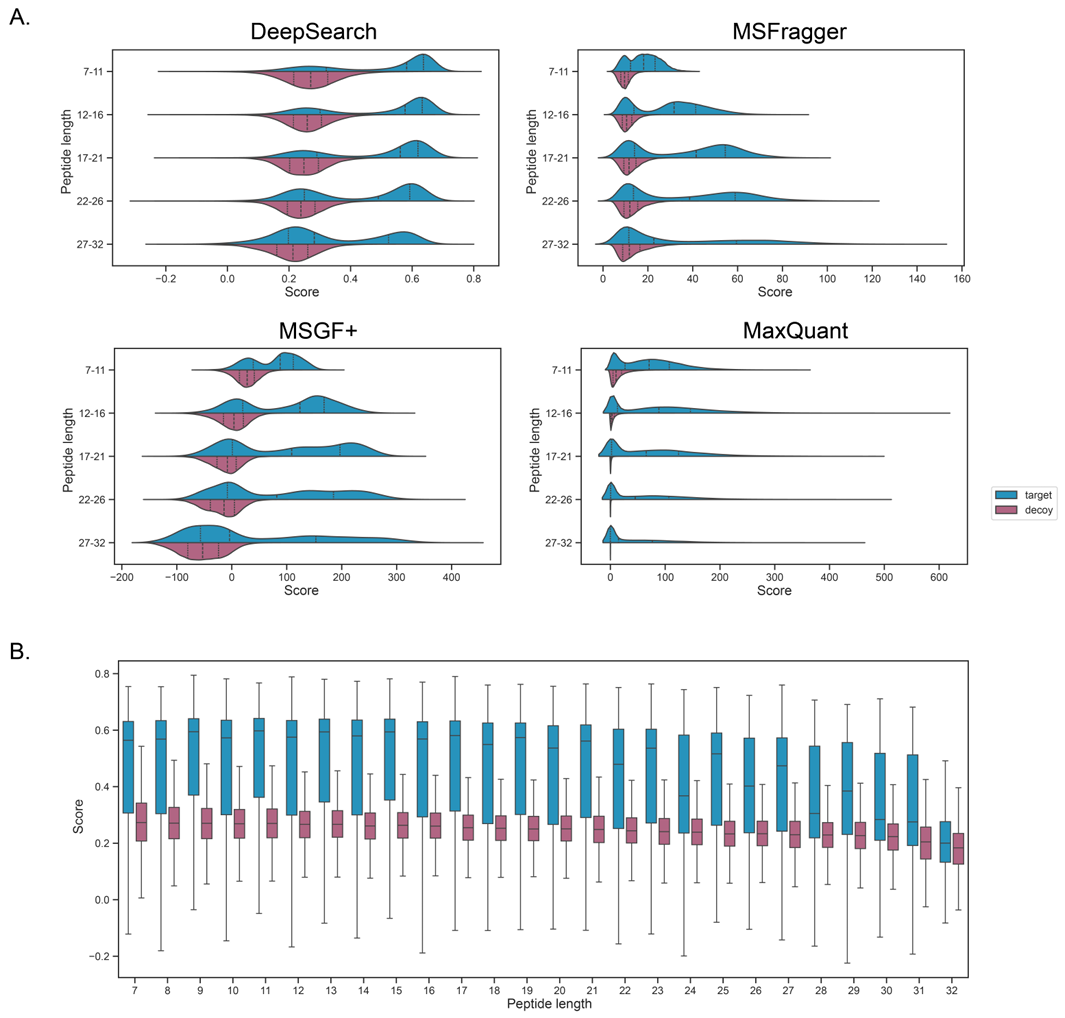
图示:搜索引擎报告了拟南芥数据集按肽长度划分的得分分布。(来源:论文)
团队在来自蛋白质组成各异的物种的多种数据集上评估了该方法。尽管 DeepSearch 只在人类光谱库上进行训练,但与所有数据集上最先进的数据库搜索引擎相比,它在 1% 伪发现率(FDR)下始终报告了相当数量的 PSM。
实验结果显示 DeepSearch 识别的大多数肽段都得到了其他搜索引擎的高度证实。这些结果表明 DeepSearch 能够准确报告肽段,并且跨物种具有稳健性。
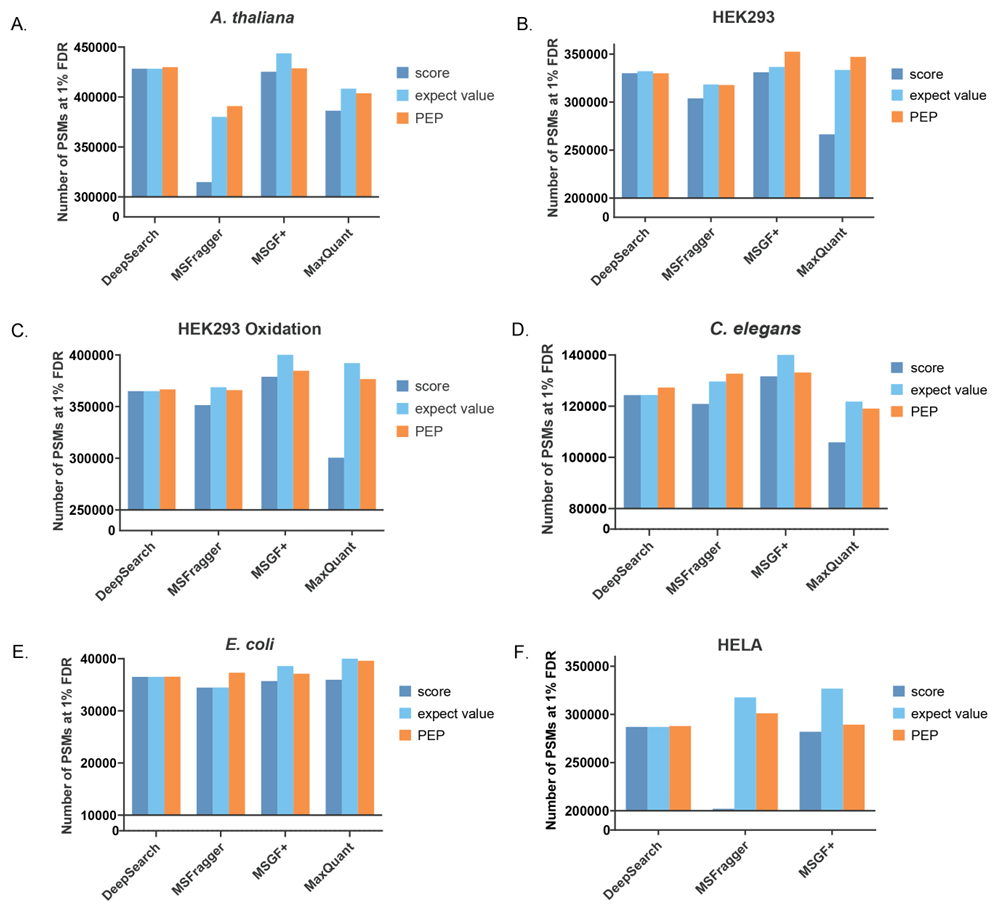
图示:多个数据集的蛋白质组范围数据集上 1% FDR 下的 PSM 数量。(来源:论文)
传统的数据库搜索引擎依赖于启发式评分函数,这可能会对某些肽组成产生偏差,并且这些搜索引擎还需要根据评分进行统计估计,以实现更高的识别率。
另一方面,DeepSearch 采用数据驱动的方法来对 PSM 进行评分。无论有没有统计模型,DeepSearch 都能保持稳定的性能,这可能与其评分方案的偏差较小有关。统计估计与目标诱饵搜索策略相结合对 PSM 质量的影响需要进一步仔细研究。
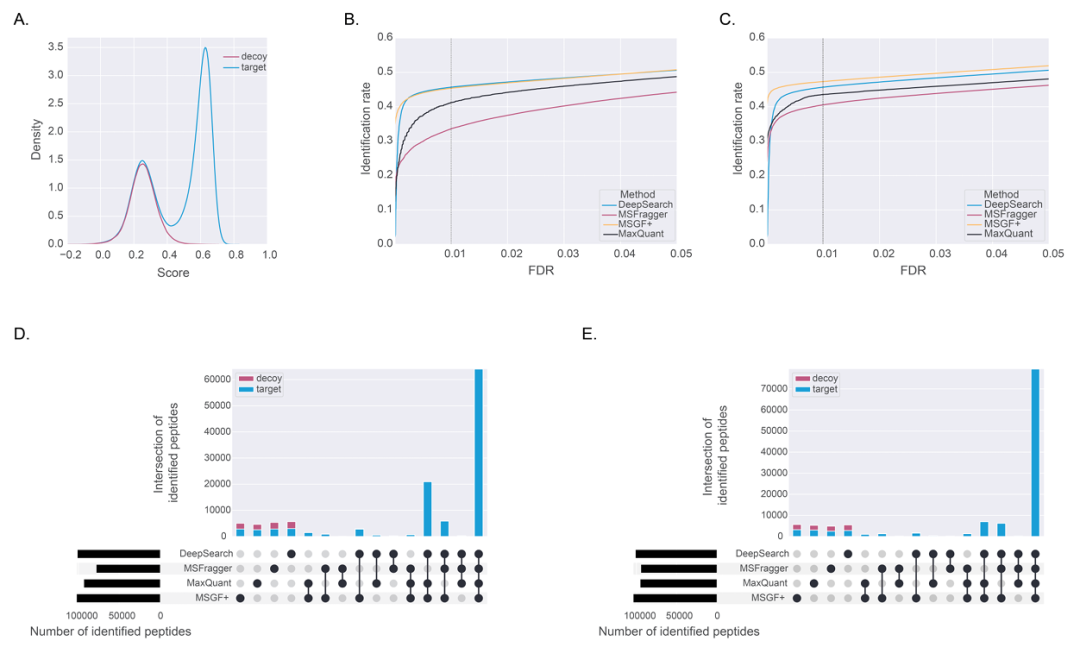
图示:拟南芥数据集的蛋白质组范围肽鉴定。(来源:论文)蛋白质组学领域中以前基于深度学习的方法通常无法进行可变翻译后修饰(PTM)分析,因为编码可变 PTM 会大幅增加标记空间。此外,将迁移学习应用于所有常见可变 PTM 的 PTM 富集数据集是不切实际的。
DeepSearch 能够报告具有磷酸化和氧化的 PTM 谱的高精度肽段。尽管如此,DeepSearch 仍需要对各种 PTM 的分析进行更多检查。
目前为止,DeepSearch 是第一种基于深度学习的方法,能够进行零样本变量 PTM 分析,而无需除 PTM 质量之外的任何先验信息。DeepSearch 通过将 PTM 移位理论谱与未修改的肽序列联合编码,绕过了标记空间限制。
论文链接:https://www.nature.com/articles/s42256-024-00960-1
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角
点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。








