
基于经验知识对反应性能进行精确预测,为高效分子设计开辟了新途径。与人类从特定数据集中总结出的反应知识相比,机器学习在大规模数据集中构建的定量结构—性能关系模型更有助于全面探索整个化学空间。
为了解决这些问题,德国哥廷根大学Xinran Chen, Lutz Ackermann教授团队以及浙江大学洪鑫团队合作在Nature Synthesis期刊上发表了题为“Integrating a multitask graph neural network with DFT calculations for site-selectivity prediction of arenes and mechanistic knowledge generation”的最新论文。
在本研究中,该团队提出了一种结合机制知情图神经网络的多任务学习流程,用于预测钌催化芳烃C–H官能团化反应中的位点选择性。该多任务架构通过并行学习多个相关任务,实现对关联知识的协同获取。嵌入的反应图则在以往的机制研究与当前的反应表示之间架起了桥梁。
在引入机制信息的基础上,所构建的多任务模型在所汇总的256条反应数据集中展现出优异的插值与外推能力,位点选择性预测的平均准确率达到0.934,标准差为0.007。该模型的预测范围涵盖了从简单芳烃到稠合芳烃的各类底物,甚至在包含14个未见样本的外部测试中也成功扩展至杂环吲哚类衍生物。
1. 实验首次结合多任务学习与机制知情图神经网络,预测钌催化芳烃C–H官能团化反应的位点选择性,得到了平均预测准确率为0.934的结果,并在标准差为0.007的情况下展现了优异的预测性能。
2. 实验通过多任务学习架构,协同学习不同任务的相关知识,实现了对钌催化C–H活化反应中位点选择性的精准预测。该模型不仅涵盖了从简单芳烃到稠合芳烃的反应底物,还扩展到杂环吲哚类衍生物的预测,且在包含14个未见样本的外部测试中也表现出良好的外推能力。
3. 实验通过机制知情的反应图,将先前的机制研究与反应表示相结合,构建了一个有效的反应表示方式,促进了位点选择性反应机制的理解,并通过密度泛函理论(DFT)计算验证了预测结果的合理性。
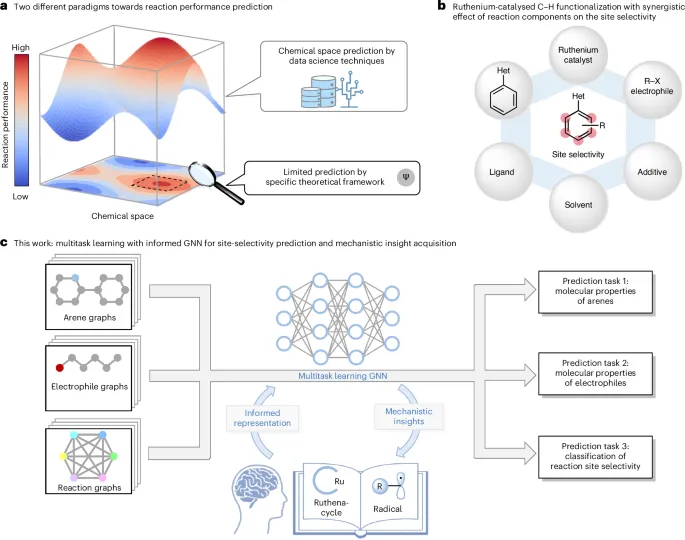
图1.通过机器学习ML算法预测反应性能。
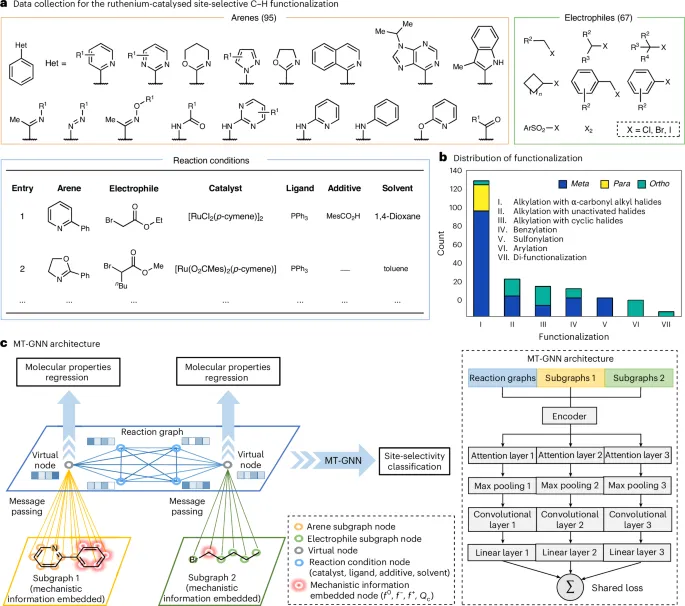
图2.多任务图神经网络multitask graph neural network ,MT-GNN的数据收集和工作流程
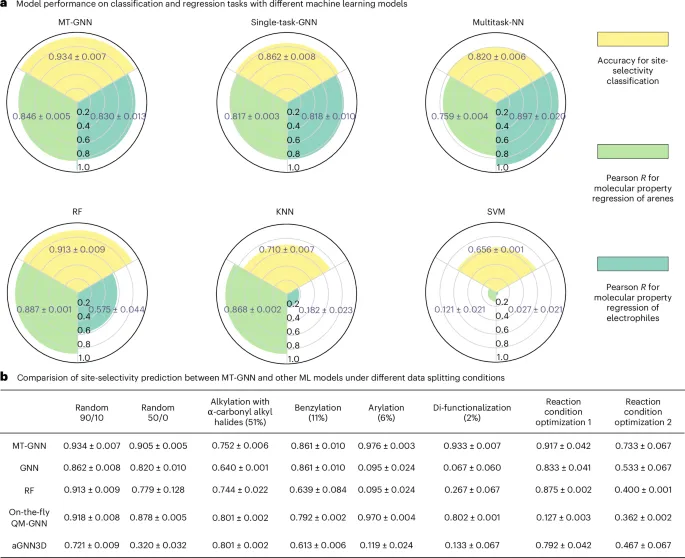
图3.多任务图神经网络MT-GNN的模型性能
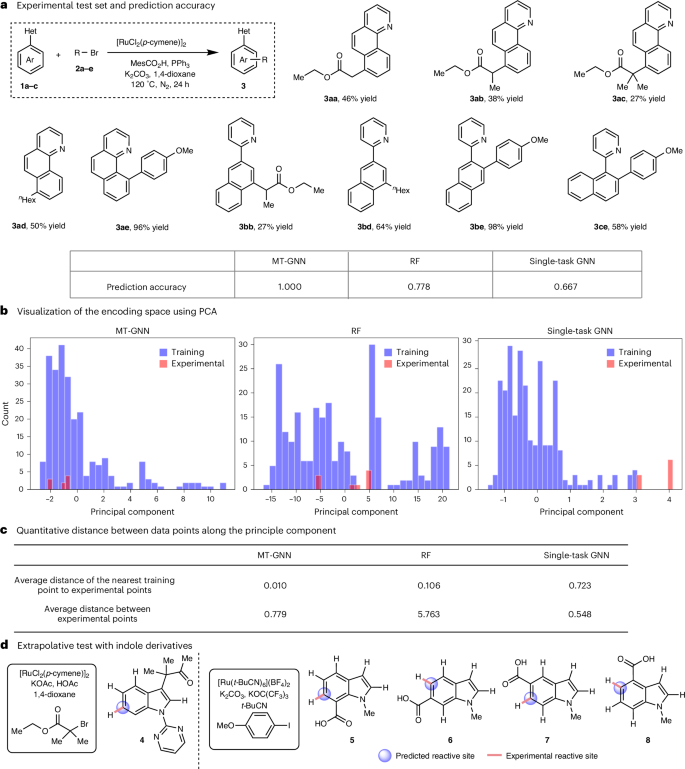
图4.多任务图神经网络MT-GNN的外推测试
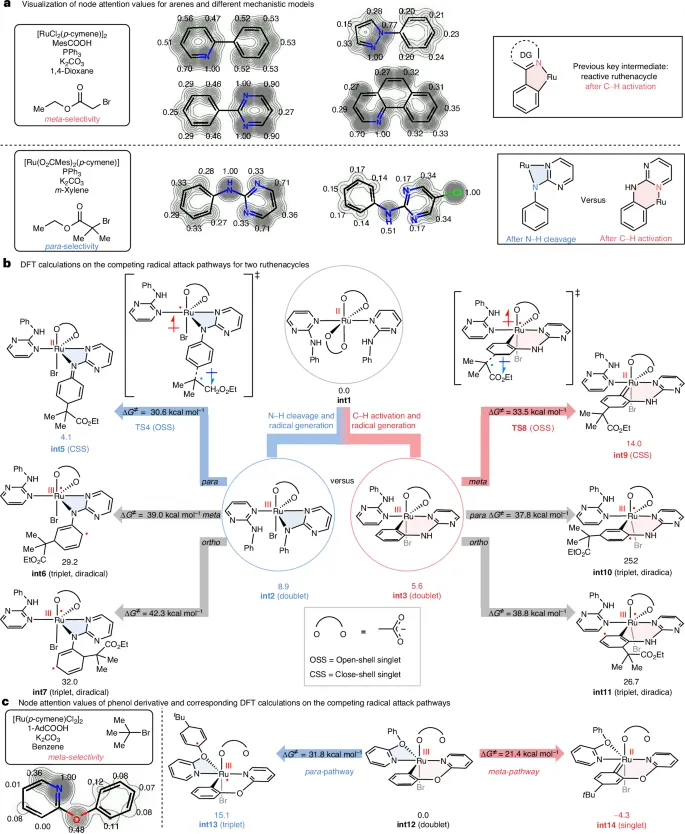
图5.多任务图神经网络MT-GNN模型的解释和DFT计算验证,从注意层提取的信息仿生机制模型
本研究展示了将机制知识融入图神经网络,并通过多任务学习策略实现高精度反应位点预测的可行性与前瞻性。首先,机器学习模型不再仅是“黑箱”工具,而是能够通过结构–性能关系的学习与可解释性机制,辅助人类提出新的反应假设;其次,机制知情的反应图设计弥合了传统理论研究与数据驱动模型之间的鸿沟,为复杂反应体系的通用预测提供了新范式;最后,MT-GNN模型不仅具备强大的预测能力,更推动了反应机理的再认知和验证过程,加速了化学从经验驱动向数据–机制融合驱动的转型。该研究为发展更加普适、高效且可解释的反应设计工具提供了重要的理论基础和技术参考,开启了人工智能辅助分子合成的新篇章。
Chen, X., Zhang, ZJ., Hong, X. et al. Integrating a multitask graph neural network with DFT calculations for site-selectivity prediction of arenes and mechanistic knowledge generation. Nat. Synth (2025). https://doi.org/10.1038/s44160-025-00770-2
免责声明:本公众号致力于分享最新科研资讯、干货资料,相关内容仅供参考学习,所有转载内容,均不代表【科学10分钟】赞同其观点,不能完全保证其真实性。如若本公众号无意侵犯媒体或个人知识产权,请联系【科学10分钟】小编:18384128522,我们将立即予以删除,谢谢。











