科研党必看!经费预存高至30%增值,更有8500+返利直接送,一次预存,全年无忧!
开发真正适用于多相催化的通用机器学习势函数仍面临巨大挑战。
2025年9月23日,上海科技大学谢闻博、胡培君在国际著名期刊Nature Catalysis发表《General reactive element-based machine learning potentials for heterogeneous catalysis》的研究论文,Changxi Yang、 Chenyu Wu、谢闻博为论文共同第一作者,谢闻博、胡培君为论文共同通讯作者。
在此,作者提出基于元素的机器学习势函数(EMLP),采用独特的“虚化学随机探索优化(REICO)”采样策略进行训练。
REICO通过遍历多样局域原子环境,构建具有代表性的原子相互作用数据集,使EMLP兼具通用性与反应性,无需预先给定结构或反应路径即可精准预测基元反应。
作者以Ag-Pd-C-H-O体系为例,构建针对Pd-Ag催化剂与含C/H/O物种相互作用的EMLP,在表面重构、覆盖度效应及溶剂环境等复杂场景中仍与密度泛函理论(DFT)定量吻合,而现有基础模型通常无法胜任。
该方法为在更大、更复杂的多相催化体系中替代DFT计算铺平道路,并提供可便捷扩展至其他催化体系及材料科学等领域的通用框架。
图1:
a) EMLP工作流程:首先用小规模随机结构及低精度DFT弛豫训练初始proto-EMLP;在REICO采样阶段,利用proto-EMLP(或全程低精度DFT)对额外随机结构进行弛豫,并通过采样技术补充稳定结构,以丰富数据集中局部原子环境(因高对称、稳定构型难以被REICO访问)。随后采用力分布直方图与SOAP描述符结合余弦相似度的多阶段筛选方案对数据集进行平衡,最终经高精度DFT计算后训练EMLP。b) Ag-Pd-C-H-O EMLP训练集的示例结构及体系尺寸分布,可见大部分计算针对约15原子体系完成。c) EMLP可对训练未涉及的任意结构进行即时计算,故可在系列代表性多相催化基准测试中替代DFT。
图2:(a)与公开数据库结构相比,本工作REICO(b)的数据多样性与覆盖度分析。横坐标为每原子能量,纵坐标为Steinhart序参量(OP,度量局域原子排布分布,任意单位),颜色(z轴)表示每个能量–OP区间内数据点数量(log10尺度/计数)。可视化结果显示,REICO数据集不仅覆盖MD数据集的大部分区域,还在能量与局域原子环境方面显著扩展。c, 通过刚性二聚体扫描进行的通用性测试,该测试未出现在训练集中,适合评估EMLP的通用性。测试包括银、钯、碳、氢、氧元素间十种自相互作用与交叉相互作用二聚体组合。DFT计算结果作为基准,与EMLP、MACE-mp与EquiformerV2预测对比;M3GNet无法完成二聚体测试。EMLP结果与DFT基准高度一致,而MACE-mp与EquiformerV2预测偏差显著。颜色标识同图1。
图3:a,由EMLP、M3GNet、EquiformerV2与MACE-mp对Ag28O22及Pd34O56团簇进行MD模拟的快照,并以DFT为参考。使用EMLP的MD全程未释放O2分子,与DFT结果一致;而其他MLP模拟中出现O2生成。b, 采用DFT、EMLP、M3GNet、EquiformerV2与MACE-mp对Ag(111)表面两个PTCDA分子进行结构弛豫,给出PTCDA不同部位碳原子与Ag(111)表面的平均距离。颜色标识同图1。
图4:每幅图均将DFT结果与EMLP、EquiformerV2、MACE-mp及M3GNet模型预测进行对比,纵坐标为以初始吸附态为基准的反应能量,过渡态结构由EMLP给出,颜色标识同图1。
图5:图a–d:依次为Pd(111)(a)、1 ML氢覆盖Pd(111)(b)、0.25 ML氢覆盖PdAg3
(111)(c)与1 ML氢覆盖PdAg(111)(d)的表面模型、DFT与EMLP能量曲线对比,以及各过渡态反应能垒差异(EMLP、EquiformerV2、MACE-mp、M3GNet)。a起始于C2H2与H共吸附,b–d起始于C2H2与表面H共吸附;颜色标识同图1。
图6:图a、c:EMLP与DFT对比的Pd(100)费托合成反应能垒图,反应路径含多步基元反应,由CO起始至C6H14生成,坐标轴标注过渡态,下方给出EMLP所得结构;b、d:各过渡态相对于DFT的绝对能量偏差(ΔE),对比EMLP、MACE-mp与EquiformerV2,颜色标识同图1。
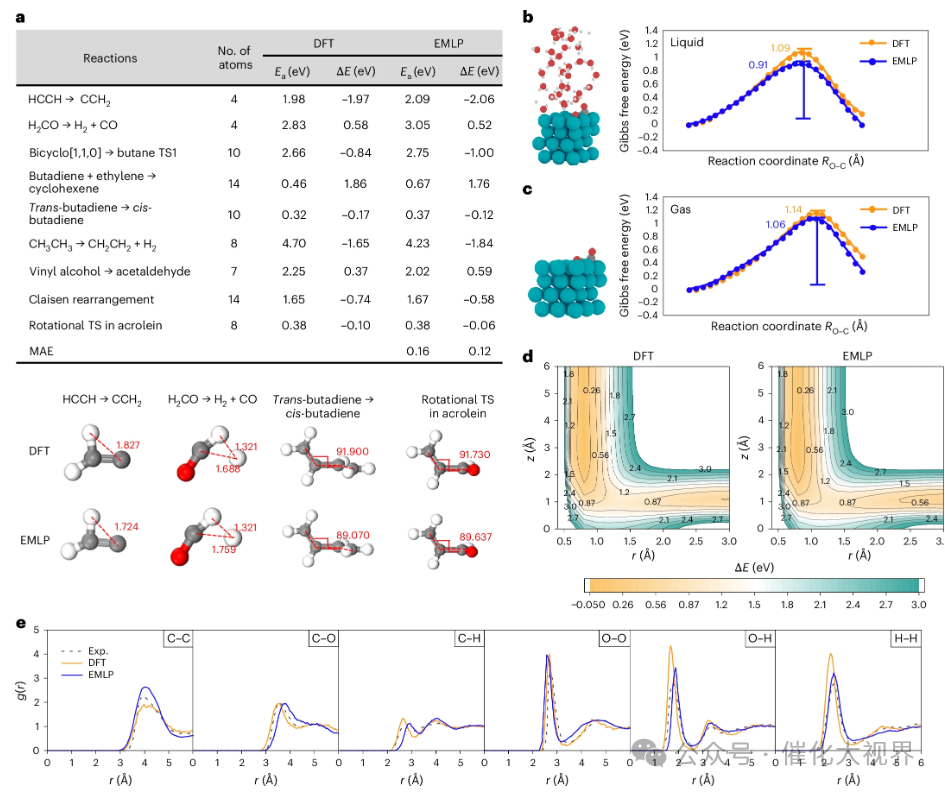
图7:a:选取Baker与Chan标准测试反应集(含部分过渡态结构对比,距离单位为Å)的DFT与EMLP活化能Ea及反应焓ΔE对比;b、c:在显式水存在(b)与无水(c)条件下,CO于Pd(111)表面氧化的自由能曲线,由DFT与EMLP结合伞形采样MD计算获得;d:H2在Ag(100)散射动力学中,固定分子中心及取向于空心位时,DFT与EMLP的二维势能面等高线图(z与r,单位Å);e:液态甲醇的径向分布函数,对比DFT与EMLP-MD结果,各面板对应O–O、O–H、H–H、C–C、C–H、C–O原子对,含H对仅取羟基氢且排除分子内对,颜色标识同图1。
本研究开发了一种基于元素间相互作用的通用机器学习势函数(EMLP),通过随机探索虚拟化学结构(REICO)策略构建训练集,无需依赖特定反应路径或结构输入,即可准确预测多元素体系中的反应能垒与结构演化。
该方法在多种催化反应(如CO氧化、乙炔加氢、费托合成)中展现出与DFT一致的高精度,具备跨体系泛化能力,为大规模复杂催化体系模拟提供了高效可靠的DFT替代方案,推动机器学习势函数从“专用”走向“通用”,在多相催化、材料设计等领域具有广阔应用前景。
General reactive element-based machine learning potentials for heterogeneous catalysis. Nat Catal. (2025). https://doi.org/10.1038/s41929-025-01398-3
#上海科技大学#催化#计算#Nature子刊#胡培君院士#谢闻博
🏅 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。🎯500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 👏👏👏









