
![]()
![]()
【研究背景】
电解质是决定电池性能、安全性与寿命极限的根本因素,然而其设计面临广阔的化学空间以及稳定性、离子传输与界面相形成之间复杂耦合与权衡的制约。本综述系统阐释了机器学习(ML)赋能全新电解质研发范式的具体路径,包括:针对溶剂、盐和添加剂开展分子层面的性质预测与逆向设计;发展可迁移的机器学习力场,将接近量子化学精度的分子动力学模拟拓展至真实混合物体系,从而架起分子结构与宏观物性之间的桥梁;以及利用可解释模型与闭环实验,实现数据高效的多目标配方优化。此外,本文还综述了物理引导的机器学习在系统级诊断中的应用,即基于早期循环数据与阻抗信号预测电池的健康状态及使用寿命。上述进展有望推动更安全、更长续航的储能技术发展。
【工作简介】
近期,华南师范大学邢丽丹教授/肖哲熙特聘研究员课题组联合美国SES AI公司首席科学家许康博士在国际顶级期刊《Chem》上发表了题为“Machine learning in electrolyte design: Accelerating discovery and multidimensional optimization for next-generation batteries”的综述文章,系统分析了机器学习在下一代电解液中所扮演的重要角色,涵盖机器学习辅助的电解液中溶剂/盐/添加剂分子优化、配方优化与电池寿命预测。该文将预测目标进行分类,对当前常见的机器学习驱动电解液设计策略进行了系统阐述,并分析了各自的利弊。相较于传统实验优化,机器学习能够大幅节约时间与经济成本,是一种绿色环保且经济高效的解决方案。然而,电解液性能受多维指标耦合权衡的制约:针对某一性质的单目标优化虽可准确提升该特定性能,但往往会出现“顾此失彼”的问题;多目标优化虽能更全面地兼顾电池的整体性能,但也面临计算成本显著上升、高质量数据获取困难等挑战。因此,如何正确协调不同因素对预测的影响,成为未来该类研究的重要挑战。相关进展共同勾勒出从数据到决策的工作流程,将计算与实验有机连接,旨在实现更安全、更长寿命的电池体系发展。华南师范大学邢丽丹教授、肖哲熙特聘研究员与SES AI公司许康博士为共同通讯作者,华南师范大学硕士研究生刘思源与美国犹他大学董登攀博士为共同第一作者。
【内容表述】
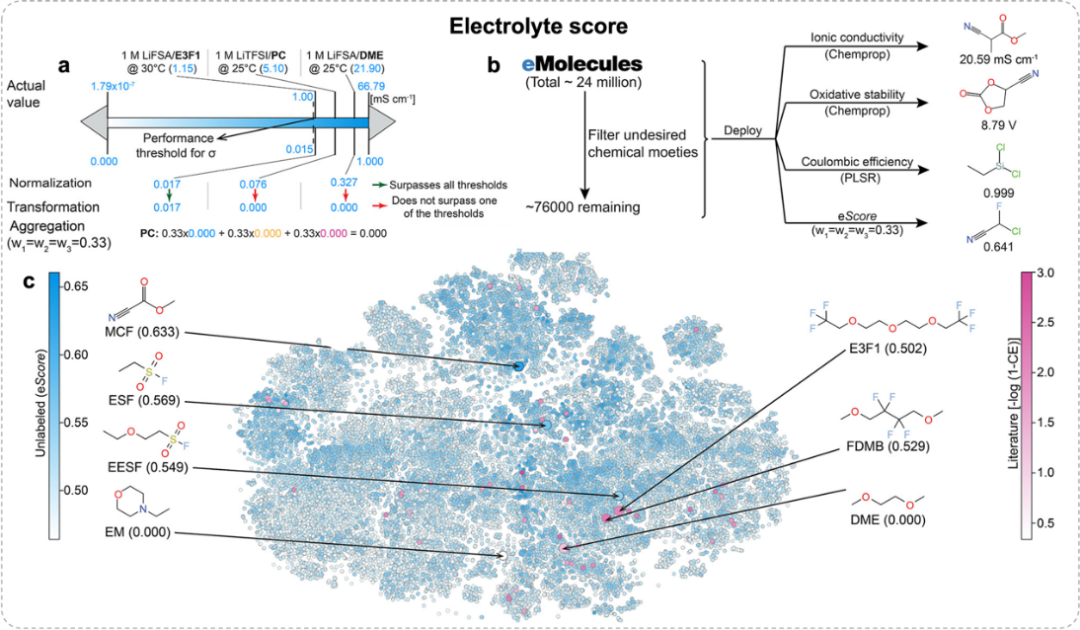 图1. 非监督学习eScore分析方法
图1. 非监督学习eScore分析方法
3.1针对电解液的分子设计:建立机器学习和化学之间的桥梁
3.1.1针对溶剂分子优化与性质预测
在机器学习驱动的分子设计中,需精确定义预测目标,以更好地从海量化学组分中快速筛选具有前景的分子。多目标贝叶斯优化及可解释人工智能(如SHAP)的引入,进一步提升了模型的透明度与实用性,有助于研究者理解分子特征与电解质性能之间的关联。与此同时,结合材料领域知识的数据库治理框架,为构建高质量、高信息量的数据集提供了系统性指导。单目标优化作为当前最为流行的预测策略,为众多研究者所采用,其预测目标涵盖氧化还原电位、结合能、配位能、库仑效率等指标,以准确表征电解液的特定性质,进而推断其溶剂化结构以及电池充放电过程中的电荷转移效率等关键参数。此外,多目标优化能够协同考量多项潜在竞争性质,从而为候选分子的排序与决策提供更全面的依据。
3.1.2从单分子到配方:面向电解质混合体系的可迁移机器学习力场
在上一节中讨论的诸多预测目标(如氧化还原描述符、稳定性指标及溶剂化相关能量)通常来源于单分子或小团簇的量子化学计算,或基于此类数据训练的机器学习代理模型。然而,电池电解质实际为凝聚态多组分液体(或凝胶),集体溶剂化、离子配对、浓度效应及动态异质性会显著影响离子输运与稳定性,导致分子级描述符难以直接映射至配方行为。因此,迫切需要发展能够清晰准确描述介观液体结构及其动力学演化过程的模拟手段。分子动力学(MD)通过积分运动方程模拟体系的时间演化,其中力场用于描述粒子间的相互作用:经典力场支持高效的大规模模拟,已广泛用于研究电解质的溶剂化与输运过程;从头算MD(AIMD)虽精度更高,但在处理大体系与长时间尺度时计算成本过高。为兼顾精度与可扩展性,机器学习力场(MLFFs)应运而生,并在电解液模拟中更具优势。MLFFs基于量子力学数据训练,能够以接近经典MD的计算成本实现近DFT级别的精度,从而在真实多组分体系相关的时空尺度上开展模拟。基于MLFF的机器学习模型不仅能够预测分子层面的氧化还原与稳定性特性,还可借助MLFF捕捉集体溶剂化及离子输运行为,并预测密度、粘度、离子电导率等宏观可测参数,从而架起从单分子到电解质配方的实用桥梁。
图2.
BAMBOO机器学习力场
3.1.3针对电解液体系中锂盐的优化
作为电解质的关键组分,锂盐(及其阴离子)在数量与结构多样性方面比溶剂和添加剂更为有限。综合调研表明,能够同时满足高溶解度、足够离子电导率及充分电化学稳定性等实际要求的盐阴离子不足15种。这一稀缺性,既源于电池对锂盐的性质有着严格且常相互冲突的要求,也源于开发具有新型稳定结构的阴离子在化学合成上面临的困难。由于锂盐的性能对其微观结构特征极为敏感,这构成了材料设计的核心挑战。目前,针对该挑战的优化主要围绕热力学性质与动力学性质两方面展开。在此背景下,机器学习技术基于上述两个关键维度,为快速筛选与发现新型高性能锂盐开辟了新的策略路径。此外,溶解度直接决定盐浓度与离子电导率,机器学习预测可辅助筛选高浓度下不发生沉淀的溶剂-盐组合。然而,实际电解质为多组分体系,溶解度受复杂的非线性耦合作用及温度的强烈影响,导致室温下训练的模型难以外推至宽温域,而多温度数据的获取成本高昂。为此,需发展物理感知描述符、高质量多温度数据集及温度信息建模策略,以实现对电解质溶解度的稳健预测。
3.1.4针对电解液添加剂的优化
成膜电解质添加剂能够在不大幅改变本体电解质配方的前提下改变界面化学性质,兼具经济性与高效性。低含量的添加剂即可显著优化SEI/CEI,抑制副反应,从而提升电池的稳定性与循环寿命。机器学习与DFT计算相结合,可有效从众多候选分子中筛选理想添加剂,成为提升电池性能的高效实用途径。
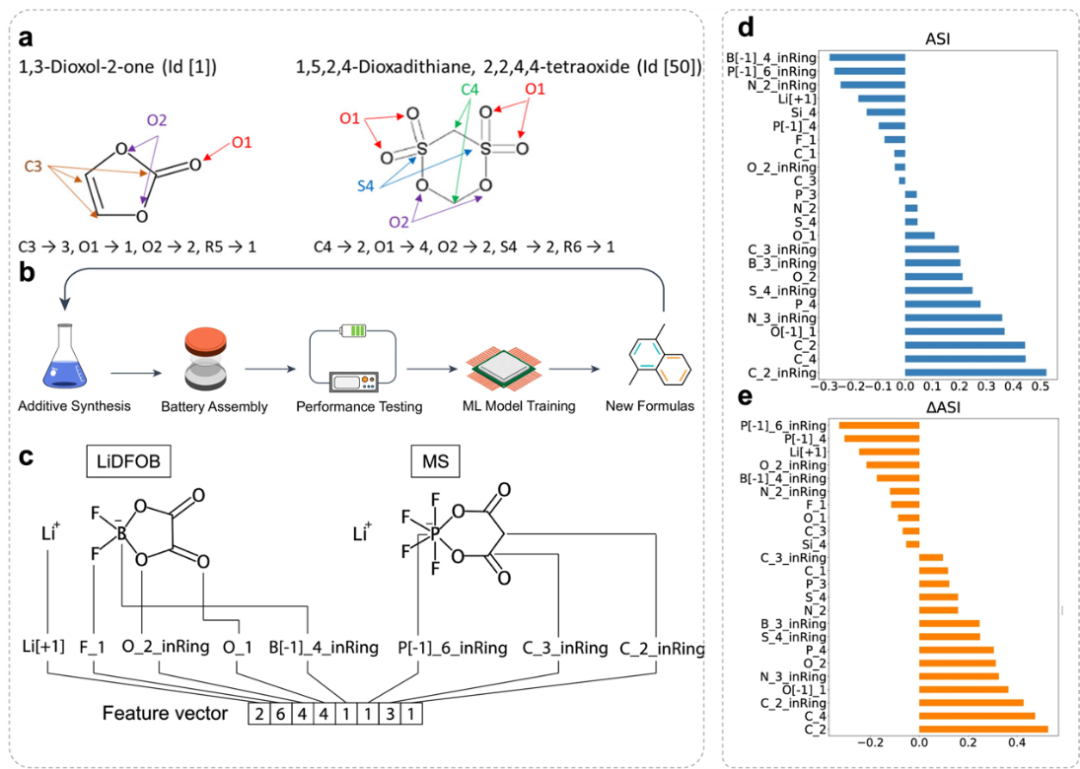 图3. 通过数据分析和特征生成的电池添加剂分子工程
图3. 通过数据分析和特征生成的电池添加剂分子工程
当前机器学习引导的添加剂研究已涵盖多种范式,包括可解释的QSPR式筛选、多目标性能建模及生成式设计。相应地,目标性质亦从氧化还原与电子描述符(如氧化/还原指标、HOMO/LUMO能级)、配位相关参数(如给体数DN)拓展至界面热力学描述符(如表面能γ、吸附能与溶剂化能)。
3.1.5基于化学特征映射的数据驱动电解质设计
化学特征映射的核心,在于将电解液分子复杂的高维信息投影至一个低维且物化意义明确的特征空间中。在此空间中,分子根据其化学与功能属性实现可视化聚类,从而建立起“结构-性质”的直观关联,为高性能电解质组分的快速初筛与理性归类提供了全新视角。基于此,研究人员融合第一性原理计算(DFT)数据库与已发表的实验/理论数据,构建了面向电解质设计的机器学习模型。该策略已成功加速了新型溶剂与添加剂的发现流程,并指导合成出数类具备优异性能(如高离子电导率、宽电化学窗口、优异界面稳定性)的候选分子。这一系列成果表明,以化学特征映射与机器学习为核心的人工智能辅助分子设计范式,正展现出成为变革性技术路径的潜力,有望系统性加速并革新各类下一代电池体系中关键电解质材料的研发进程。
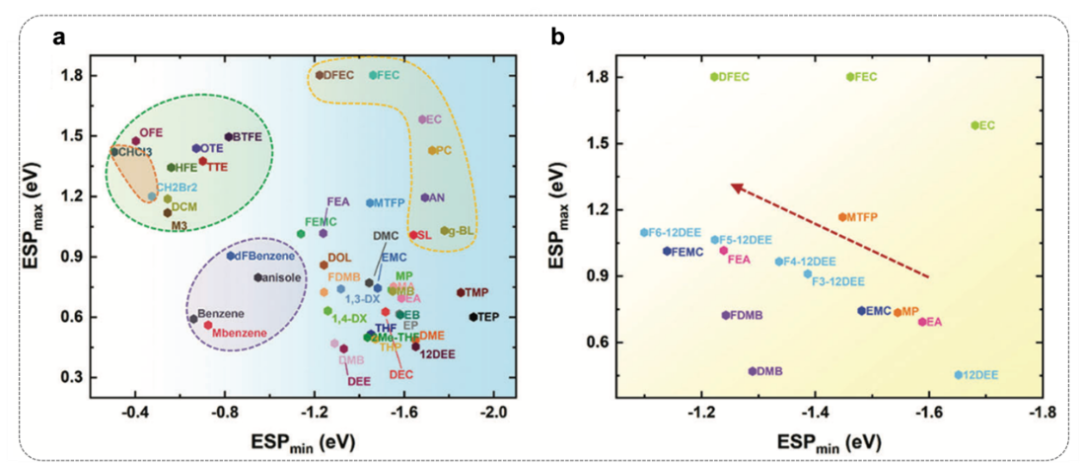 图4. 化学特征映射
图4. 化学特征映射
3.1.6基于机器学习的电解质分解与界面相演化机理研究
电池界面相(包括SEI和CEI)的形成对电池的寿命、稳定性和可逆性至关重要。在机理层面,界面相的生长源自一个由众多耦合与竞争反应构成的庞大网络,涵盖还原、氧化、键断裂与重组等过程。仅依靠传统的量子化学方法难以解析这一网络,原因在于反应路径的组合数量巨大、凝聚相环境的影响不可忽略,且体系对界面条件高度敏感。当前,机器学习结合DFT与AIMD能够准确预测溶剂分子在电极表面的吸附能及氧化还原反应电位等性质,从而建立分子性质与CEI/SEI成膜机理及其性质之间的关联。此外,机器学习对过渡态的精确识别同样有助于深入探究界面反应机理。
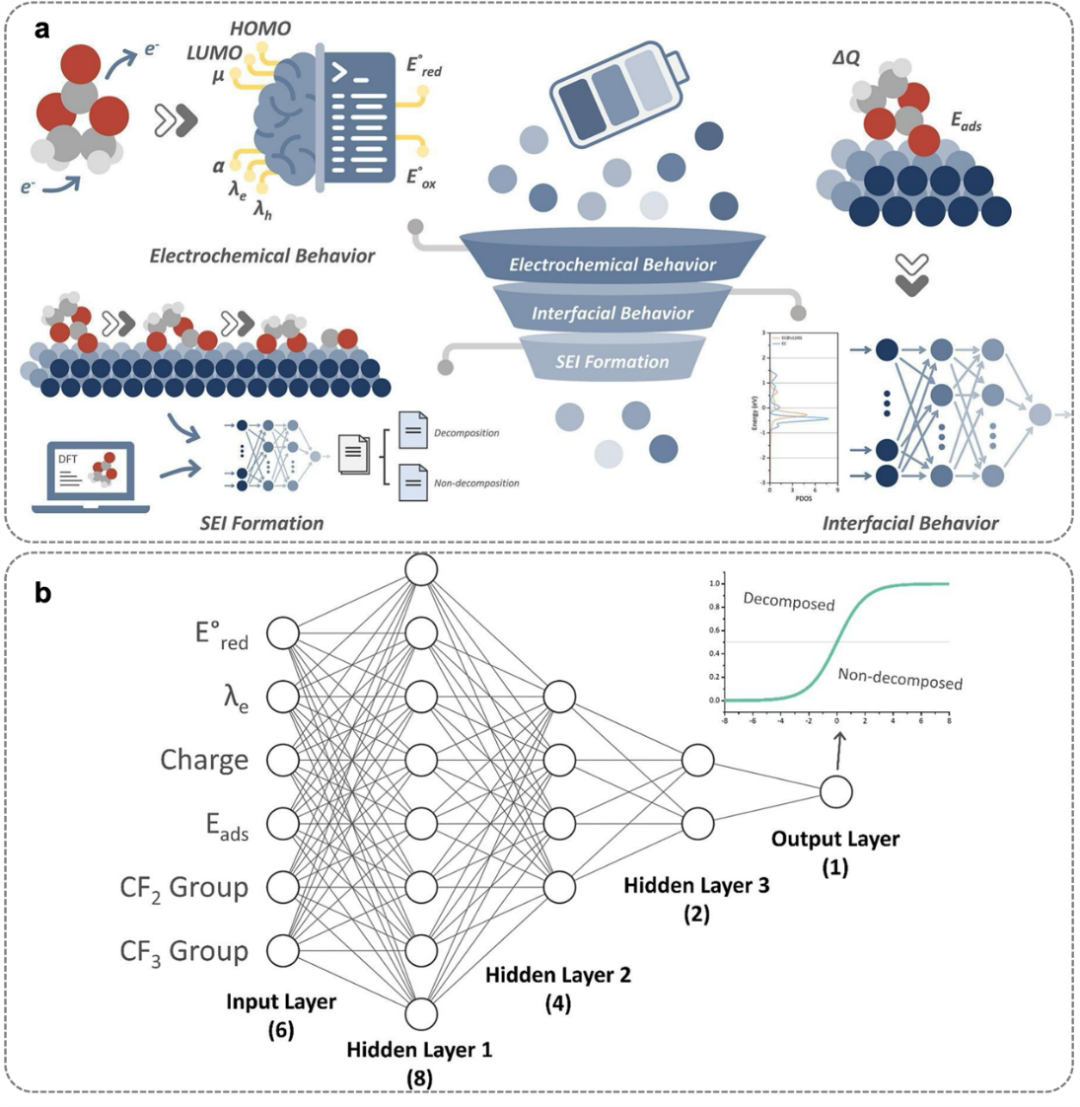 图5. DNN机制工作流程
图5. DNN机制工作流程
3.1.7面向机器学习引导优化的参数化设计
机器学习引导的材料优化中,一项先决条件在于对优化“参数”的精心设计,即可控设计变量的选择、目标函数及其权重的定义,以及物理约束的引入。参数化步骤通常决定了贝叶斯优化或其他搜索策略能否高效识别高性能候选方案,尤其在多个目标相互竞争的条件下。针对下一代电解质,类似的参数化可在多个层面加以定义,包括分子选择与配比、盐浓度、添加剂种类与用量、聚合物结构及加工条件,同时目标指标涵盖传输性能、稳定性、界面相容性与安全性等。
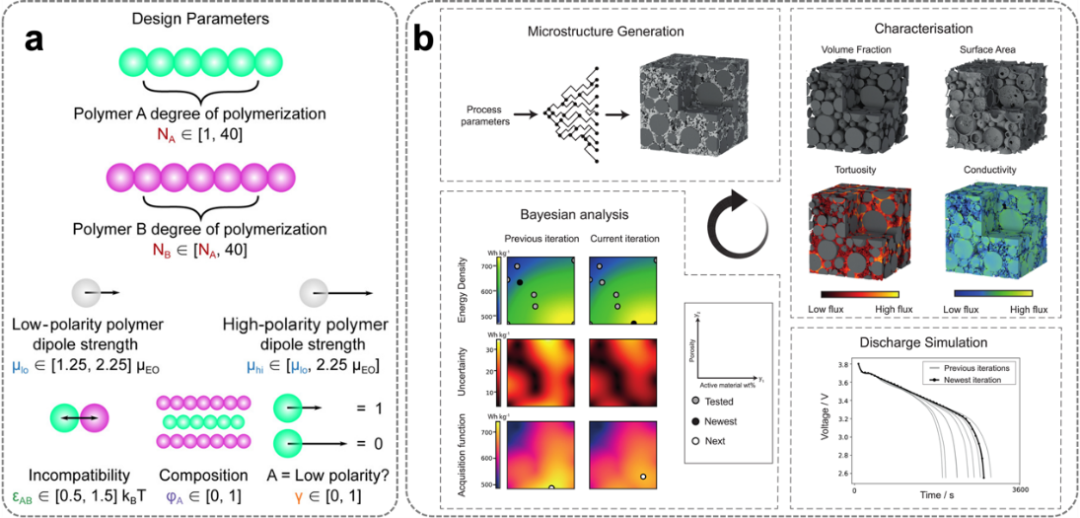 图6. 基于贝叶斯优化的 Stockmayer 模型参数及其电解质设计应用
图6. 基于贝叶斯优化的 Stockmayer 模型参数及其电解质设计应用
3.2 电解液配方优化
在机器学习加速电解质研发中,库仑效率(CE)因直接反映电化学可逆性,成为配方优化的核心目标。有研究基于对数CE模型与元素描述符,揭示溶剂分子中的氧元素占比降低是提升CE的关键,据此设计的新型醚类电解质实现了99.70%的超高CE。另有研究采用贝叶斯优化与帕累托前沿对CE与过电位进行多目标平衡,优化后的局部高浓度电解质使自生成负极电池容量保持率由45%提升至55%,CE达99.39%。然而,CE受测量条件与数据偏差等因素制约,单一目标优化易忽视离子传输与安全性的协同关系,由此催生了多属性建模策略,例如通过同时预测溶剂化结构参数来解析CE机制。机器人自主实验平台“Otto”与集成AI平台“Uni-Electrolyte”融合逆合成分析,正在构筑从单分子设计到多目标系统优化的全链条智能范式,为更高性能、更长寿命的下一代电解质开发提供强劲动力。
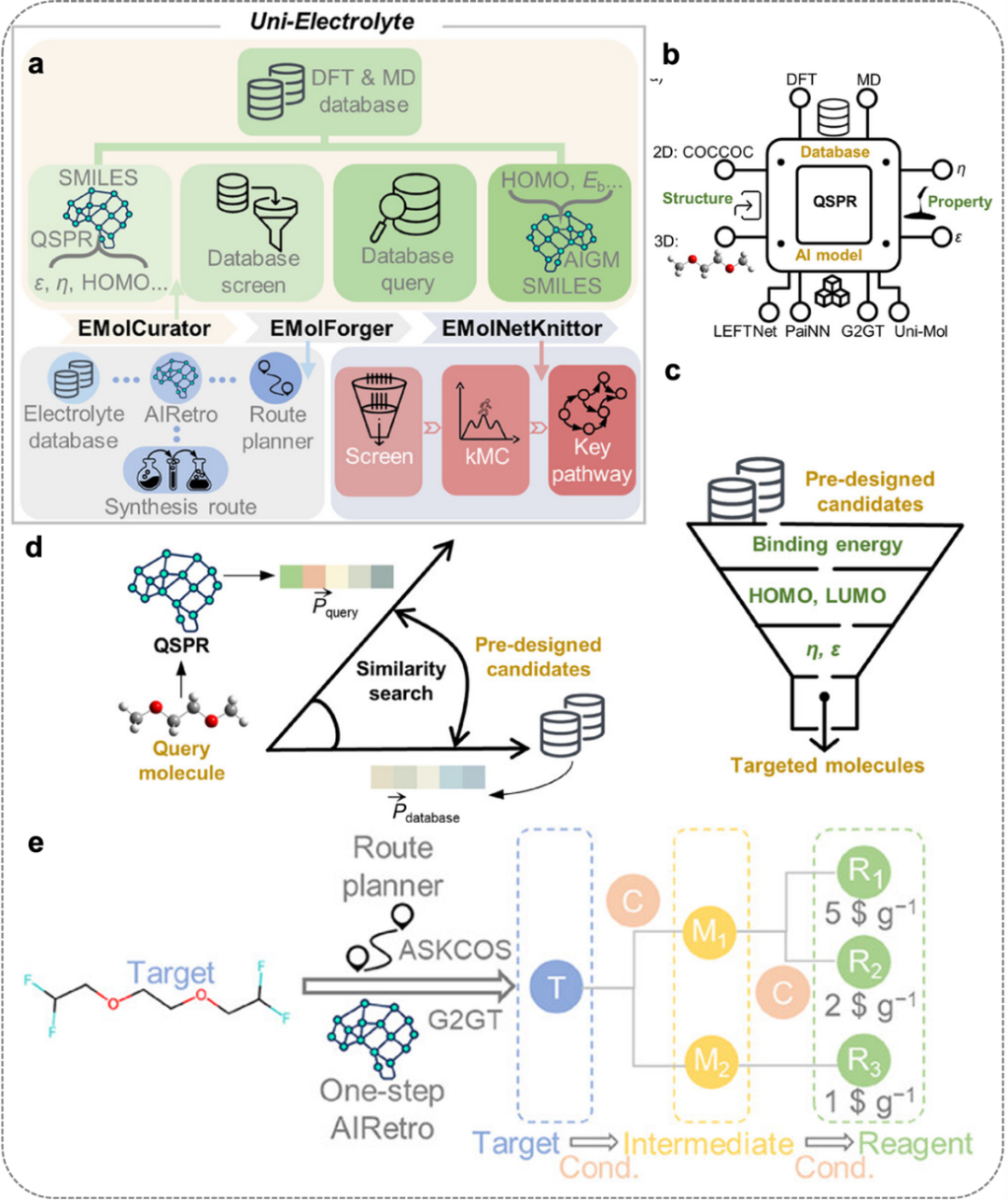 图7. 从分子设计到逆合成的电解质发现自主计算流水线
图7. 从分子设计到逆合成的电解质发现自主计算流水线
3.3 电池寿命与健康状况预测
传统电池寿命测试耗时长、成本高,而机器学习正开辟一条数据驱动的新路径,使研究者仅基于早期循环或部分运行数据即可精准推断健康状态与剩余寿命。借助SA-LSTM及BatLiNet等深度架构,可从放电电压曲线中提取关键特征,在初期实现高精度的容量衰减与循环寿命预测;有研究表明,融入老化相关特征后,预测误差可压缩至7.5%。为减轻实验负担,“发现学习”框架无需寿命标签即可推断全新电池设计的循环表现。在工程实用性方面,有工作仅基于部分电压区间提取统计、热力学及动力学特征,使健康状态估算精度提升超过60%;另有研究构建物理信息神经网络,仅利用充电前期数据即可高精度估算健康状态,平均绝对百分比误差低至0.65%,且在不同化学体系中展现出优良的迁移性。同时,富含频域信息的电化学阻抗谱亦被用于剩余寿命预测;针对电动垂直起降飞行器等新场景,相关模型实现了基于任务的健康管理,测试R²值约0.98,展现出良好的实用前景。
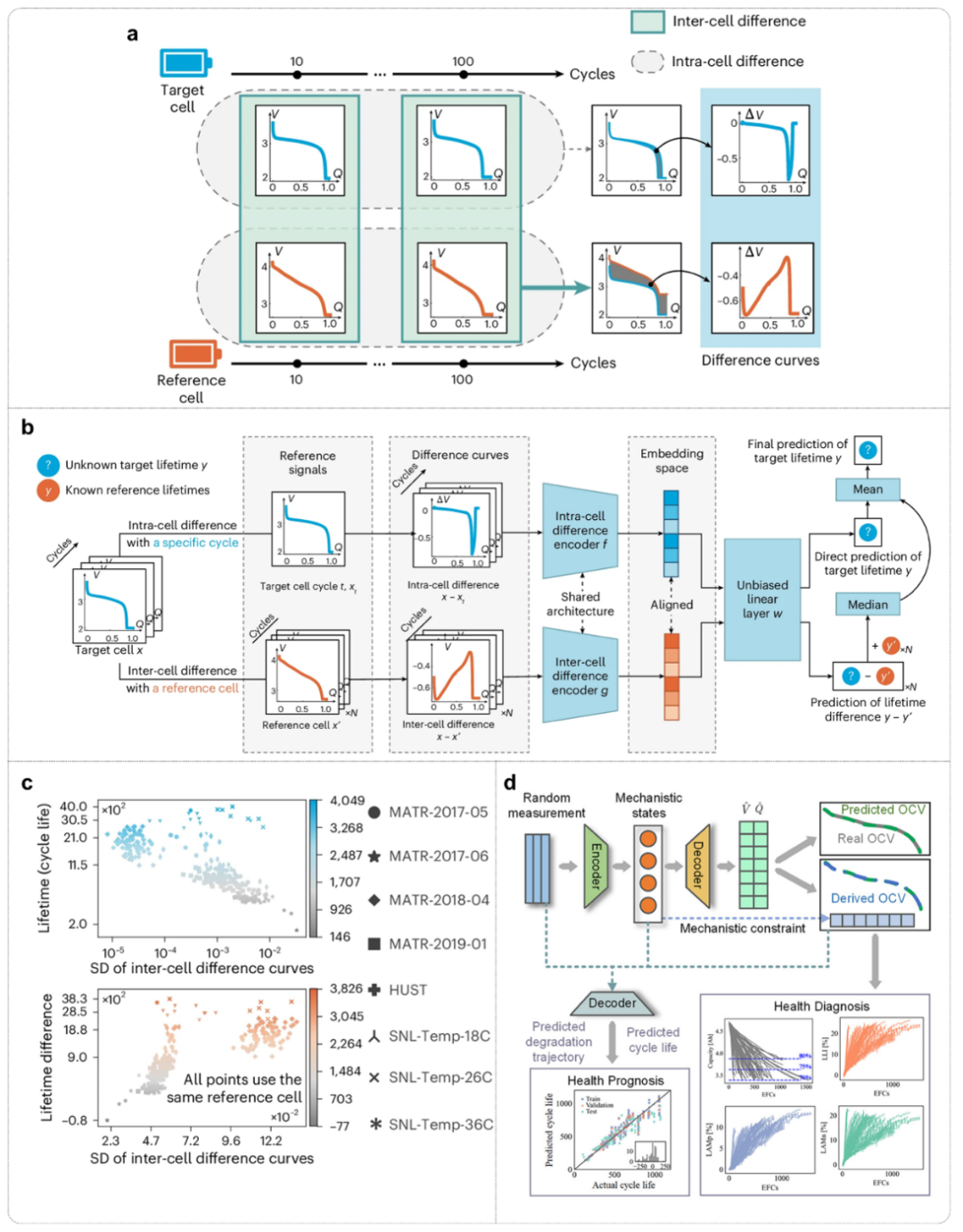 图8. 融合特征工程与预测模型的多模态电池数据挖掘
图8. 融合特征工程与预测模型的多模态电池数据挖掘
【结论展望】
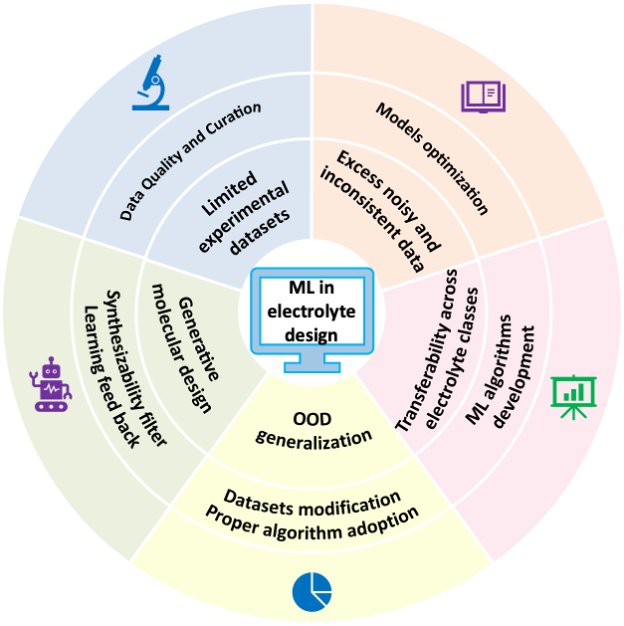 图9. 机器学习驱动电解液优化的挑战与新机遇
图9. 机器学习驱动电解液优化的挑战与新机遇
本综述系统总结了机器学习在先进电解质开发中的最新进展。内容从分子层面的优化出发,涵盖了溶剂、盐和添加剂的筛选、SEI设计,以及化学特征映射与参数优化等策略。上述方法共同提升了模型的预测精度,加速了新型电解质的发现。随后,针对数据驱动的配方优化进行了深入讨论,从分子、原子及热力学角度为改善电池性能提供了经济可行的路径。在电池寿命与健康管理方面,详细探讨了如何通过关联电解质组成、充放电制度与循环寿命进行预测,并延伸至延长电池寿命的分子驱动策略。随着大语言模型(LLM)的兴起,AI辅助电解质研究获得了更广泛的关注。基于领域知识训练的LLM有望整合并生成超出训练语料范围的新知,但其“幻觉”问题,尤其在涉及化学准确性时,仍是严重隐患。未来需从算法、领域增强技术及高质量数据集三方面提升LLM的科学严谨性。总体而言,机器学习已成为变革性的研究手段,本综述聚焦于液态电解质设计、物化性质与电化学反应的前向预测以及寿命预估。上述技术与LLM相结合,有望构建未来的智能体平台,大幅减少实验开销与开发周期,加速电池研发与产业应用。
基于当前研究进展,作者对未来机器学习驱动的研究指出了当前困境与未来发展方向:
(i)有限实验数据集及其对模型训练的影响:当前,由于实验成本高昂且周期漫长,尤其在材料信息学与电池研究等领域,实验数据稀缺仍是一项根本性挑战。这一限制严重制约了稳健机器学习模型的训练,数据不足常导致泛化能力差及过拟合。为缓解上述问题,研究者通常采用K折交叉验证以增强模型稳健性,并在数据集受限条件下实现更可靠的性能评估。然而,真实的实验基准与算法生成的数据(包括合成模拟输出或增广数据集)之间始终存在差异。例如,尽管DFT等计算方法可扩充数据池,但往往难以完整反映真实实验条件的全部复杂性。此类不匹配最终推动了对全面、高保真数据库日益增长的需求。同时,对算法与模型进行适当优化亦是应对数据有限问题的可行解决方案。
(ii)多模态数据融合中的噪声与不一致数据:融合来自多种表征技术(如SEM、XRD、TEM等)的数据对于全面分析材料性能至关重要。然而,由于不同研究中测量条件与样品制备流程的差异,以及技术本身局限引入的人为伪影,导致数据中混杂着显著的噪声与不一致性,使得多源数据融合过程极为复杂。破坏性制备技术,例如常与SEM/TEM联用的聚焦离子束(FIB)铣削,会对样品造成不可逆损伤。特别是离子铣削等非原位表征技术,在截面制备过程中会去除关键且敏感的材料层,从而改变原始微观结构,并阻碍对同一区域的后续多模态分析。
(iii)跨电解质类别的可迁移性:电解质模型通常在特定类别(如固态电解质)内表现优异,但对其他类别的泛化能力有限。这一限制主要源于不同类别间主导离子输运机制的本质差异。当前多数研究集中于单一类型的电解质,原因在于开发跨多类别的统一模型需要庞大的数据集,这会增加过拟合风险,使模型倾向于记忆训练数据模式而难以外推至未知体系。为克服这一挑战,研究者正探索迁移学习策略,即利用从某一电解质类别获取的知识提升对其他类别的预测精度,旨在构建具有广泛适用性的模型,从而加速先进电池的开发。此外,在特定情况下采用先进且合适的算法亦可克服上述困难。
(iv)分布外泛化:数据驱动的机器学习模型难以推广至其训练域之外,这一局限被称为分布外(OOD)泛化问题。例如,在碳酸酯类电解质上训练的模型应用于氟化醚溶剂、新型盐或多价体系时往往失效,原因在于关键的物理机制可能未被包含于训练集中。尽管迁移学习与物理信息机器学习可提供一定缓解,鲁棒的分布外泛化仍是实现新电解质化学体系预测性发现的主要障碍。为应对这一挑战,对数据集进行合理优化、扩充与划分,并结合恰当的算法,可有效降低分布外情形所带来的负面影响。
(v)生成式分子设计:扩散模型与化学语言模型等生成式模型能够从头探索化学空间,并以目标性质为条件生成候选溶剂、盐及聚合物。然而,许多生成所得的分子在合成上难以实现,或在电化学条件下稳定性不足,且往往仅满足单一目标性质而无法兼顾其他。因此,将生成式设计与可合成性过滤器及主动学习反馈相结合,对于将分子生成转化为实用的电解质发现至关重要。
【文献详情】
原文题目:Machine Learning in Electrolyte Design: Accelerating Discovery and Multidimensional Optimization for Next-Generation Batteries
发表期刊:Chem
DOI:10.1016/j.chempr.2026.103024
作者:Siyuan Liu1, Dengpan Dong1, Zhexi Xiao*, Dmitry Bedrov, Kang Xu*, Lidan Xing*, Weishan Li
原文链接:https://doi.org/10.1016/j.chempr.2026.103024
【作者简介】
通讯作者:肖哲熙特聘研究员、许康博士、邢丽丹教授
第一作者:刘思源 董登攀
作者单位:华南师范大学,犹他大学,SES AI公司
肖哲熙,华南师范大学化学学院特聘研究员,硕士生导师。2020年6月毕业于清华大学化学工程系并取得工学博士学位。2024年9月加入华南师范大学化学学院电化学研究所邢丽丹教授团队。在Chem, Adv. Mater., Energy Environ. Sci., ACS Energy Lett., Adv. Funct. Mater., Nano-Micro Lett.等期刊上发表论文20篇,以第一/通讯作者发表15篇,第一发明人授权发明专利7项。主持国家自然科学基金面上、青年基金、广东省自然科学基金以及校企联合攻关等项目,入选广东省科协青年科技人才培育计划。
邢丽丹,华南师范大学教授,博士生导师,电化学研究所所长。长期致力于研究电极/电解液界面双电层中,电解液组分、构型与所形成界面膜性质的构效关系,发展了系列电解液调控新策略和功能添加剂设计方法,构筑了高稳定的界相,大幅提高了锂/钠离子电池的综合性能。代表性成果发表在Nat. Chem., Joule, Natl. Sci. Rev., J. Am. Chem. Soc. 和Chem等期刊上。
许康,美国材料研究学会(MRS)会士、电化学学会(ECS)会士、陆军研究院(ARL)退休院士,目前担任位于马萨诸塞州波士顿的SES AI公司首席科学家。从事电解质材料和界面化学研究30余年,代表性研究包括新型电解质材料(溶剂、锂盐、添加剂、非易燃电解质)的设计和合成,以及基础界面和相间过程化学,对超浓缩水相电解质、电池化学和相间研究具有开创性意义。









