
催化剂发现常被限制在某一类材料内部:研究单原子催化剂就用单原子数据,研究钙钛矿氧化物就用钙钛矿数据。这样做稳妥,但也让模型难以吸收跨材料规律,更难预测一个过去没有训练过的新材料类别。
这篇 Nature Materials 提出 crossbreeding neural network,简称 CBNN。作者从碳载单原子催化剂和块体钙钛矿氧化物两个实验数据集中抽取共同描述符,再用统一模型预测一个新的目标类别:负载在钙钛矿氧化物上的单原子催化剂。
这类问题的难点在于,不同材料族的结构输入、活性来源和实验指标并不完全相同。作者通过自动统计筛选和自然语言分析找到共享活性相关特征,再把表面单原子分支和体相氧化物分支耦合,让模型能在不同材料域之间迁移信息。
成果简介
模型筛选出五个关键共描述符,包括氧化态、离子半径、离子电子数、电负性和配位数。用这些描述符联合训练后,CBNN 对已有材料的 OER 过电位趋势具有较好相关性,并能预测 SACs on perovskite oxides 这一未训练类别。
实验验证显示,模型预测的单金属候选趋势与实际结果吻合。进一步筛选 8008 个多金属 SAC 候选后,最佳 MM/CPCF 由 W、Mo、Ru 和 Rh 低含量单原子共同负载在 CPCF 上,内禀 OER 过电位为 349 mV at 10 mA cm-2 oxide,优于训练组和目标组中的既有候选。
可解释机器学习进一步把描述符重要性和表面原子贡献联系起来,指出氧化态和配位环境等因素如何影响活性趋势。文章的价值在于证明跨材料模型不只是做插值,而是可以提出真实可合成、可验证的新催化剂组合。
研究亮点
亮点一:把碳载单原子和钙钛矿氧化物两个实验数据域连接起来,突破单一材料族内训练的限制。
亮点二:CBNN 不只预测单金属趋势,还筛出多金属 MM/CPCF,并经实验验证达到更低 OER 过电位。
亮点三:可解释分析把共描述符、表面图像激活和活性趋势联系起来,让模型输出更接近可理解的材料设计规则。
配图精析
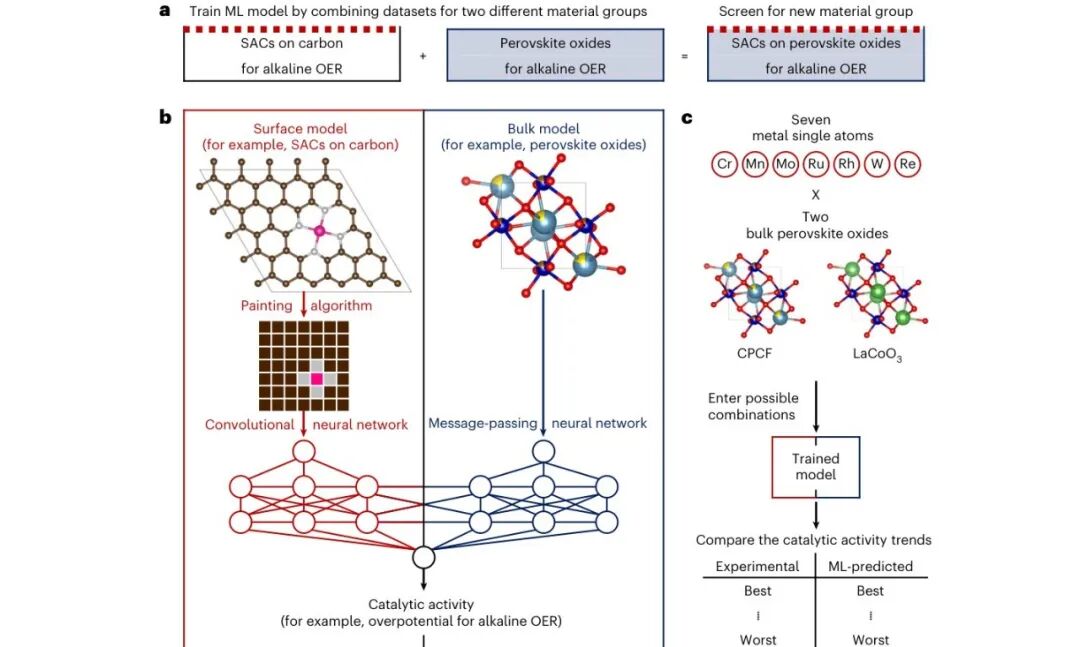
图1:CBNN 如何把两个材料族“交叉育种”
示意图展示了模型思想:一个分支学习碳载 SAC 的表面结构,另一个分支学习块体钙钛矿氧化物结构,随后在目标材料 SACs on perovskite oxides 中同时启用两个分支。这样做相当于把单原子位点和氧化物载体的经验放进同一个预测框架。
这个框架的巧妙之处在于,它并不要求两个数据集长得完全一样,而是通过共享描述符把它们投影到可沟通的特征空间。对实验数据稀缺的催化领域来说,这种跨域借力比单纯扩大同类数据集更现实。
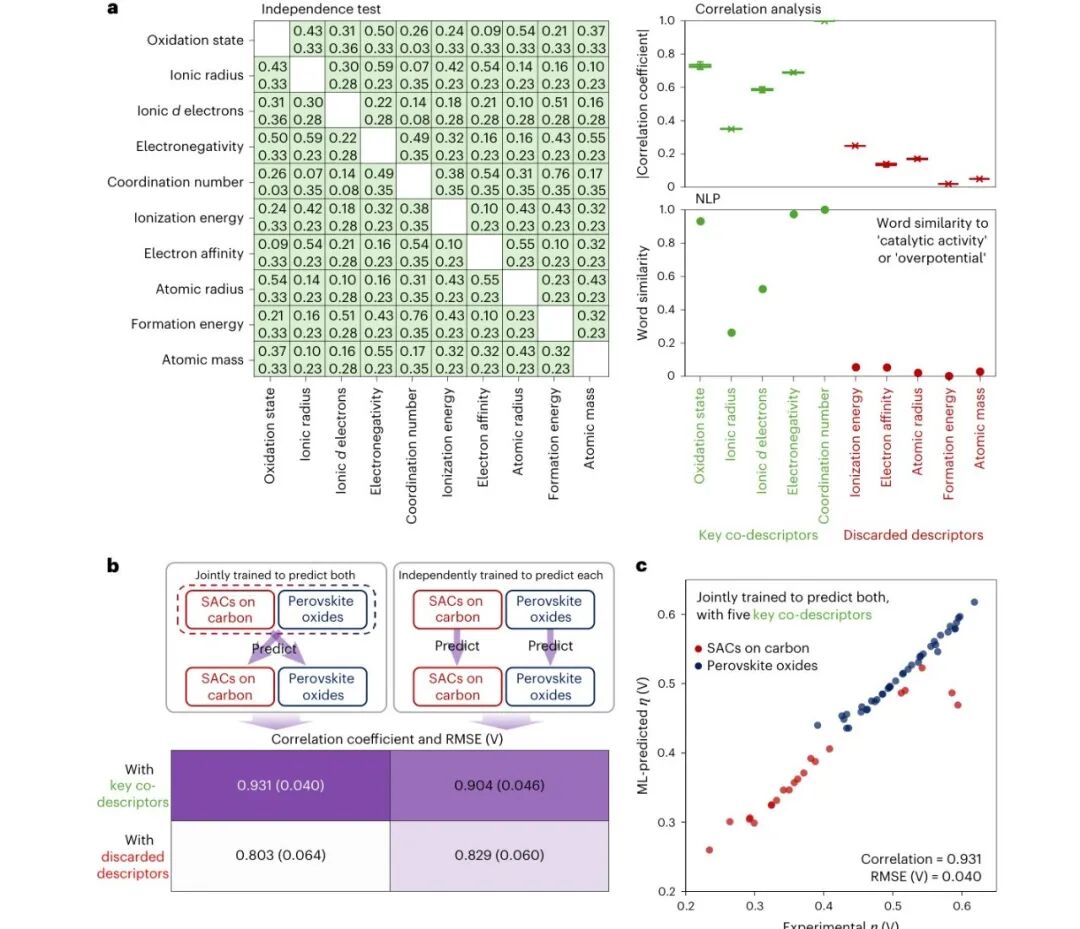
图2:共描述符筛选和跨材料训练效果
热图和筛选流程说明,作者不是任意拼接特征,而是通过独立性检验、相关性分析和语义相似度筛出共享描述符。联合训练后,模型在两个材料域中都能较好对应实验过电位,说明这些共描述符确实捕捉了跨材料的 OER 活性信息。
这一步很关键,因为机器学习催化研究最怕把无关特征堆进模型,得到看似高分但不可解释的相关性。作者用统计和文本信息双重筛选,让模型输入更接近化学直觉,也减少了跨材料迁移时的噪声。
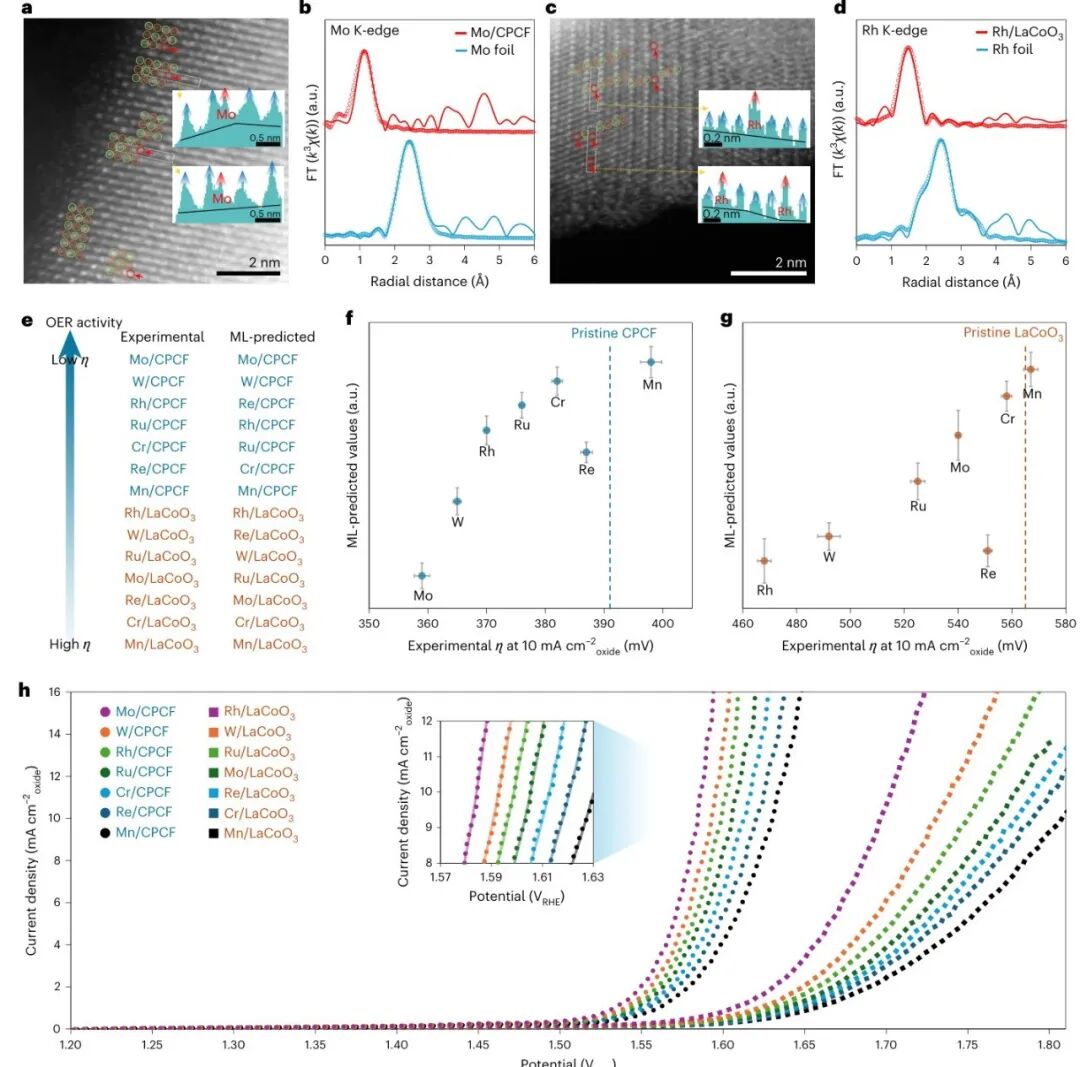
图3:单金属候选的结构表征和预测验证
Cs-STEM 与 EXAFS 证明 Mo/CPCF、Rh/LaCoO3 等样品中金属以单原子形式存在,没有形成金属颗粒。右侧实验和预测趋势的对比说明,CBNN 能识别不同金属在不同氧化物载体上的相对活性,为后续多金属组合筛选打下基础。
实验验证让模型从“算法演示”变成材料发现工具。如果预测趋势只停留在计算表格里,可信度有限;这里通过合成、显微和电化学同时验证,说明模型确实抓住了载体和金属位点之间的活性规律。
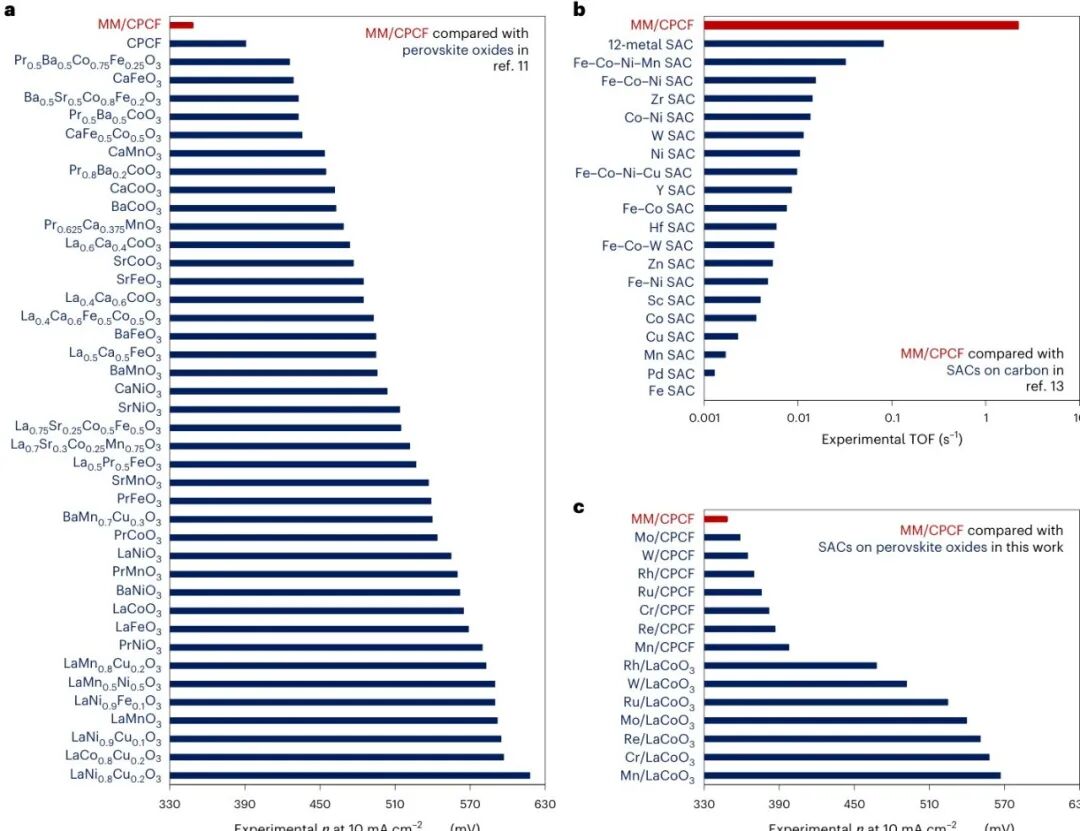
图4:MM/CPCF 超越三个材料组的既有候选
这张排名图是文章最直接的结果。MM/CPCF 在 bulk perovskite oxides、SACs on carbon 和 SACs on perovskite oxides 三类材料中都排到前列,实验过电位达到 349 mV。它说明模型筛出的多金属组合不是纸面最优,而是可以在实验中保持结构并表现出优势。
更有意思的是,MM/CPCF 不是简单选择一个最强单金属,而是用低含量 W、Mo、Ru、Rh 的组合实现协同。模型在这里承担了人眼很难完成的任务:从数千种比例组合里找到值得合成的一组。
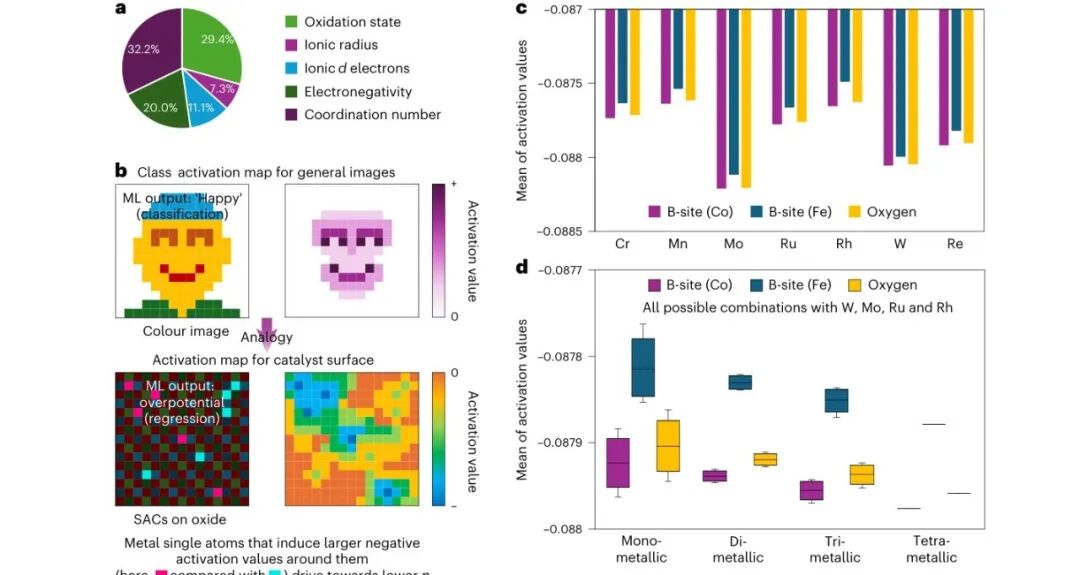
图5:可解释模型揭示哪些特征驱动活性
可解释性图把黑箱输出拆成可读信号:氧化态、离子半径、离子电子数、电负性和配位数对模型判断有不同权重,表面图像激活还指出哪些区域更影响预测。对于催化剂发现来说,这一步能把模型建议转化成下一轮材料设计语言。
这种解释不一定等同于完整机理,但它能告诉实验者下一轮该优先调哪些变量。相比只给一个候选清单,可解释模型更像一个能持续迭代的假设生成器,适合和高通量合成、表征平台结合。
总结展望
这项工作给机器学习催化剂发现提供了很清晰的方向:不要只把模型困在单一材料家族里,而是寻找能跨域迁移的描述符和结构表示。后续挑战在于扩大高质量实验数据、纳入稳定性指标,并让模型同时考虑活性、成本和可合成性。
文献信息
文献参考:Cross-material catalyst discovery via deep learning. Nature Materials, 2026.
原文链接:https://doi.org/10.1038/s41563-026-02622-6
点击阅读原文直达文献~

声明:更多内容请参考原文,如有侵权,后台联系编辑删除。







