近年来,人工智能与高通量计算正在加速材料研发模式的转变,但对于复杂掺杂体系而言,仅依赖传统平均组分描述符往往难以准确反映局域原子环境对材料性能的作用机制。本研究提出了一种融合高通量第一性原理计算与机器学习的研究范式,以K0.5Na0.5NbO3(KNN)基陶瓷为对象,构建了包含240个原子的掺杂模型,并结合高通量DFT计算获得了300组掺杂KNN材料数据集,涵盖KNN在2 mol%掺杂及空位缺陷下的结构稳定性(形成能)和电子结构(带隙、总能)等关键性质。研究中不仅考虑了不同元素在A位、B位及O
位的掺杂行为,还进一步分析了缺陷形成与带隙调控机制。针对传统机器学习模型依赖全局组分描述符、忽视局域掺杂环境对性能决定性影响的局限,设计了一套能够表征“掺杂原子局域环境”的结构特征,通过引入局域配位权重,设计了两类特征:组分特征(CE)与结构特征(SE),其中SE通过空间加权方式量化掺杂原子周围邻近原子(K/Na/Nb/O)的贡献,显式捕捉局域配位环境,有效增强了模型对低浓度掺杂效应的识别能力。此外,研究还结合特征重要性分析,提出了原子尺度的物理可解释的设计规则,例如,局域原子体积、核磁频率及第三电离能等关键参数直接关联体系稳定性与带隙调控,揭示了影响KNN稳定性与电子结构的关键因素,为后续高性能压电与光电陶瓷材料的定向设计提供了新方法与可靠数据库支撑。
 图1. 基于高通量计算与机器学习的KNN材料设计流程。
图1. 基于高通量计算与机器学习的KNN材料设计流程。
 图2. 高通量计算筛选的KNN掺杂元素周期表;橙色背景表示引入KNN体系的掺杂元素,带有实验标识的元素代表对应掺杂体系已完成了实验制备。
图2. 高通量计算筛选的KNN掺杂元素周期表;橙色背景表示引入KNN体系的掺杂元素,带有实验标识的元素代表对应掺杂体系已完成了实验制备。
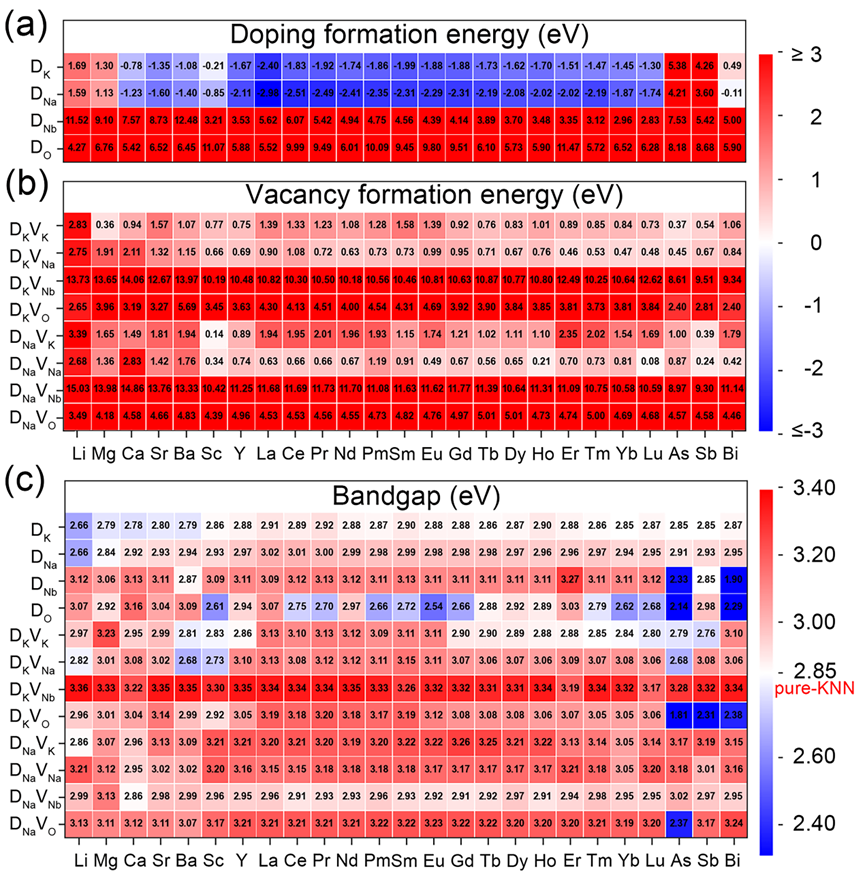
图3. 高通量计算得到的KNN体系掺杂形成能、空位形成能及带隙分布热图:(a)Ef,(b)Hf,(c)Eg。
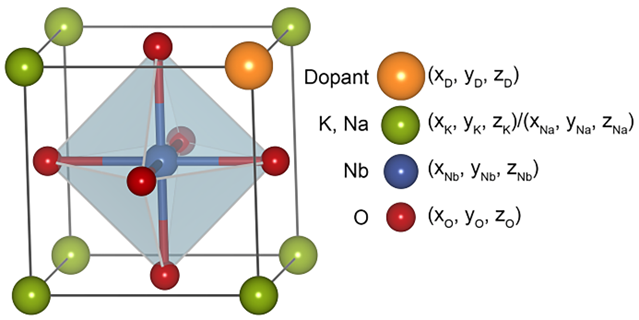
图4. 掺杂原子局域环境结构特征示意。
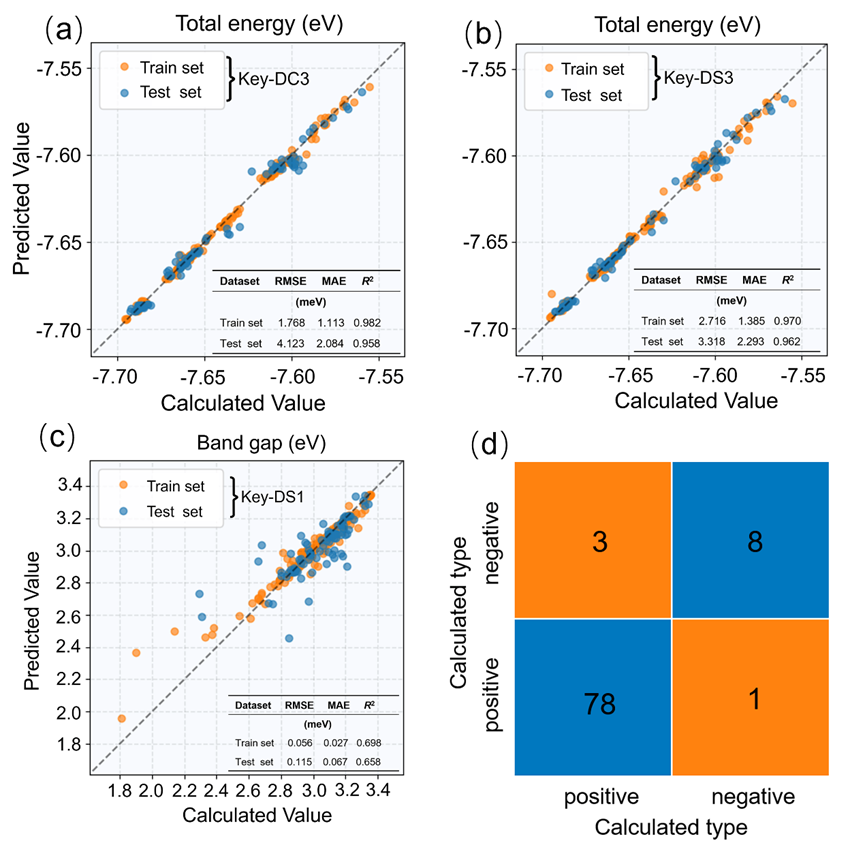
图5. 随机森林模型在关键特征集上的性能评估结果:(a)key‐DC3上预测总能的结果对比图,(b)key‐DS3上预测总能的结果对比图;(c)key‐DS1上预测带隙的结果对比图;(d)key‐DS1’上预测带隙变化的分类混淆矩阵。
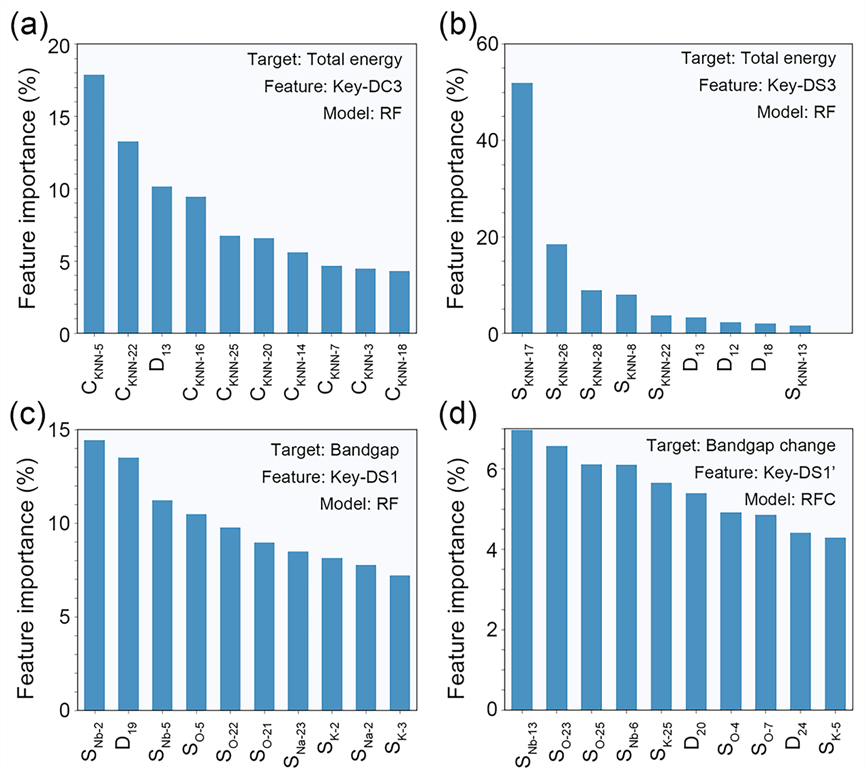
图6. 随机森林(RF)模型前10位特征的重要性评分:(a)Key-DC3上预测总能的特征贡献评分,(b)Key-DS3上预测总能的特征贡献评分,(c)Key-DS1上预测带隙的特征贡献评分,(d)Key-DS1上预测带隙变化的特征贡献评分。










