今天给大家介绍Bioinformatics期刊的一篇文章——MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction。文章提出了MusiteDeep,这是第一个用于预测一般磷酸化和激酶特异性磷酸化位点的深度学习框架。MusiteDeep将原始序列数据作为输入,并使用具有新型二维注意机制的卷积神经网络。MusiteDeep在一般磷酸化位点预测和激酶特异性磷酸化位点预测方面都展现了良好的性能。
1、介绍
翻译后修饰(PTM)通常是指在蛋白质上添加共价官能团,如磷酸化、乙酰化、甲基化和泛素化,是增加蛋白质组学多样性的关键机制。磷酸化指在蛋白质或其他类型分子上,加入一个磷酸基团,苏氨酸、丝氨酸、酪氨酸等氨基酸残基能被磷酸化,研究最多的PTM是丝氨酸和苏氨酸上的磷酸化。据估计,三分之一的哺乳动物蛋白质可能被磷酸化。这种修饰在细胞内信号转导中起着至关重要的作用,并参与调节细胞周期的分化、转化、肽激素反应等,因此,了解和鉴定磷酸化对细胞生物学和医学研究至关重要。磷酸化预测工具分为两类:第一类工具预测可以磷酸化的位点;第二类工具预测可以被特定激酶磷酸化的位点。现有的方法大多有一个共同的策略,可以概括为两个主要步骤:(1)从原始序列或其他数据中提取特征,在机器学习中称为“特征工程”;(2)利用提取的特征选择一种机器学习算法进行训练和预测。除了机器学习算法的不同,预测的成功在很大程度上取决于有效的特征提取和蛋白质表示。2、数据集
对于一般的磷酸化位点预测,数据集来自UniProt/Swiss-Prot收集的磷酸化数据,丝氨酸(S)、苏氨酸(T)或酪氨酸(Y)上的磷酸化位点被标记为阳性数据,其他则标记为阴性数据。数据集如表1所示。
对于激酶特异性磷酸化位点预测,蛋白质序列数据集也是来自UniProt/Swiss-Prot收集的数据,数据的注释是从RegPhos中提取的,对于每个激酶家族,训练一个特定的预测模型,只有被特定激酶家族注释的位点标为阳性数据,其他则标注为阴性数据,数据集如表2所示。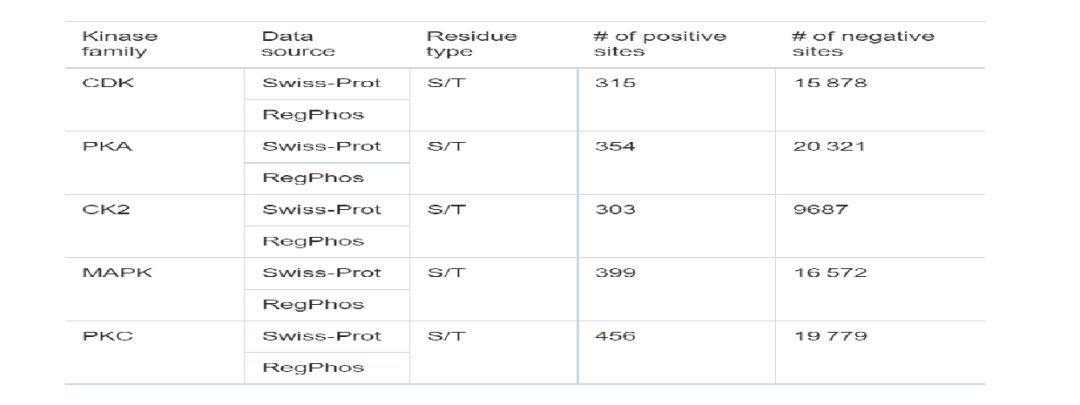
3、Musitedeep框架

图1.musitedeep架构
输入层是以预测位点为中心的33个残基的蛋白片段的one-hot编码。采用多层CNN作为特征提取器,但不使用池化层。多层CNN的最后一个隐藏状态被复制两次,一个直接输入注意机制(attention-1),另一个进行反定位,然后输入另一个注意机制(attention-2)。将这两种注意机制的输出结果结合起来,并输入到全连接的神经网络层中,最后一层是一个具有softmax输出的单一神经网络层。4、一般磷酸化位点预测
给定蛋白质序列,一般磷酸化位点预测可以预测被丝氨酸、苏氨酸或酪氨酸磷酸化的位点。它可以表述为一个二元分类问题,即每个潜在的位点都可以划分为磷酸化位点或非磷酸化位点。与其他传统的磷酸化预测工具相比,本文方法只将原始序列作为输入,给定一个蛋白质序列,提取一个以预测位点为中心的33个残基的肽。本文选择蛋白质片段长度为33(预测位点及其两侧各16个残基),蛋白质片段采用one-hot编码,即蛋白质序列中氨基酸对应的指数值为1,其他位置值为0。对于未知或非标准的氨基酸,例如缩写为“X”的氨基酸,所有位置均为0.05。由于有20种常见的氨基酸,所以K被设置为20种。然而,当一个潜在磷酸化位点的左侧或右侧部分不够16个氨基酸时,使用破折号(-),按照额外的氨基酸处理。因此,Musitedeep实际上使用了one-of-21编码方式。在MusiteDeep的深度学习架构中,多层CNN将一个输入的蛋白质序列编码为一个固定的二维隐藏状态(如图1所示).然后,将二维隐藏状态输入注意力机制1,再将二维隐藏状态反位输入注意力机制2。将这两种注意机制的输出结果结合起来,并输入到全连接的神经网络层中。5、激酶特异性磷酸化位点预测
对于激酶特异性磷酸化位点预测的挑战是如何在训练数据集较小的情况下训练一个高精度模型。根据RegPhos数据库,只有激酶家族CDK、PKA、CK2、MAPK和PKC有超过100个已知的磷酸化位点。为了与其他激酶特异性预测工具进行比较,本文重点研究了这五大激酶家族。由于训练数据集很小,很容易对训练数据进行过拟合。为了解决激酶特异性磷酸化位点预测的小样本问题,本文在一般磷酸化数据上训练了一个基网,然后将除基网的最后一层输出层外的整个网络层转移到激酶特异性模型中。最后,使用激酶特异性数据对整个网络进行了微调,通过这种方式,激酶特异性模型从一般特征表示中学习,解决了过拟合问题。
6、性能比较
6.1评价MusiteDeep在一般磷酸化位点预测中的性能
为了评估 MusiteDeep 、Musite以及其他深度学习架构的性能,使用了五重交叉验证。对于所有方法,都使用相同的训练集和相同的测试集。图2绘制了五项测试的平均ROC和精密召回曲线。它表明,所有深度学习架构的表现都优于基于特征提取的工具Musite,MusiteDeep的性能优于其他深度学习架构。
图2.一般磷酸化位点预测性能比较
6.2与其他激酶特异性磷酸化位点预测工具的比较
将MusiteDeep与几个众所周知的激酶特异性预测工具进行了比较(其中包括Musite、NetPhos3.1、GPS2.0和GPS3.0),采用CDK、PKA、CK2、MAPK和PKC五大激酶家进行比较。为了评估五个激酶家族中每个家族的性能,进行了五倍交叉验证测试,五分之四的数据用于训练MusiteDeep,其余五分之一的数据被用作MusiteDeep的测试集。对于其他工具,通过使用他们预先训练的模型进行预测,图3中绘制了激酶家族CDK和PKA的ROC曲线和精密召回曲线,表明MusiteDeep在激酶特异性磷酸化位点预测性能优于其他工具。

图3.激酶特异性位点预测性能比较(CDK、PKA激酶家族)
7、结论及讨论
MusiteDeep作为一般磷酸化和激酶特异性磷酸化位点预测的新方法,将原始序列数据作为输入,而不使用其他工具来生成特征。MusiteDeep已被证明在基准数据集上的表现明显优于其他一些知名工具。本文认为Musitedeep的卓越性能主要归功于以下三个方面:(1)本文提出的深度学习架构比基于特征工程的方法更好地捕获了与磷酸化相关的潜在序列模式;(2)引导集成深度学习方法以无偏的方式利用所有的阴性数据;(3)双重注意机制进一步提高了性能。据所知,这是深度学习方法在一般或激酶特异性磷酸化位点预测中的首次应用。除了在预训练模型中进行磷酸化位点预测外,MusiteDeep还提供定制的模型训练,使用户能够使用自己的数据训练模型。








