大家好,本周推荐一篇发表在Nature Biotechnology上的论文,题目是“Prediction of protein–ligand binding affinity from sequencing data with interpretable machine learning”,通讯作者是美国哥伦比亚大学的Harmen J. Bussemaker教授。该课题组的研究方向是通过整合基因组序列、转录因子结合和基因表达数据等信息来理解基因调控网络。

关键的细胞过程,如基因调控和信号转导,依赖于序列特异性分子识别来引导组成蛋白优先与特定的核酸或多肽配体相互作用。这种序列识别的强度和特异性通常跨越数量级,甚至弱配体也能发挥功能。因此,对这些分子网络进行全面、定量的剖面序列识别至关重要。
大规模并行测序大大提高了序列识别的速度。特别是高通量测序方法与随机配体库的体外选择相结合,已成为无偏分子相互作用分析的有力工具。这包括用于转录因子(transcription factor, TF)和RNA结合蛋白的SELEX方法,以及用于蛋白酶和T细胞受体的蛋白显示方法。由于这些分析中使用的随机配体池极其复杂(而且大多数序列很少被观察到),机器学习方法已成为将测序数据合成到识别模型的关键,该模型对任何序列如何被识别进行编码。
近年来,已有多种方法利用深度学习、概率混合模型或高维嵌入来分析TF和DNA的结合数据。然而,尽管蛋白质相互作用在生物物理参数(如解离常数,Kd)方面得到了最严格的量化,但这些方法大多侧重于将序列分类为结合或自由或分配非生物物理结合评分。虽然已经开发了一些生物物理方法,但它们仅限于估计TF的相对Kd值,不能系统地模拟多轮SELEX富集。此外,虽然已经开发了新的分析方法来分析除直接序列识别外的体内效应,但目前还没有计算方法可以将这样的补充实验合成到一个统一的结合模型中,从而捕捉辅助因子和DNA甲基化的影响。
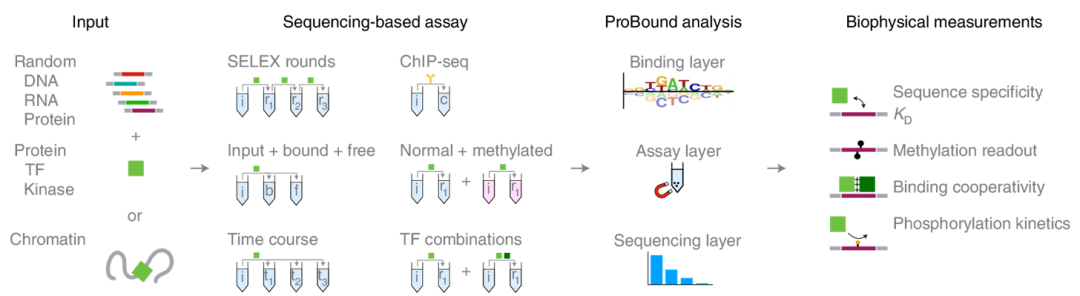
在这项研究中,作者使用一种灵活的机器学习框架ProBound解决了这些问题,该框架能够通过综合广泛的测序数据来学习生物物理可解释的模型。ProBound 使用三层对多文库测序数据进行系统建模: 结合层利用序列识别模型预测结合自由能或酶的效率;分析层对生成文库和预测所有配体频率的选择步骤进行编码;测序层对测序过程中文库的随机抽样进行建模。这些层结合在一个似然函数中,优化该似然函数来推断识别模型。ProBound可以探测以前无法探测的生物物理参数,使测序分析的发展成为可能。为了说明这一点,作者引入了Kd-seq(使用输入、结合和非结合SELEX分数来测量Kd的绝对值)和kinase -seq(使用多时间点蛋白显示分析来描述激酶底物的特异性)。更广泛地说,作者的结果说明了经典的生化分析(通常使用多个分数、时间点或浓度)如何通过测序和原则性的机器学习来进行前所未有的生物物理测量。
本文作者:YSL
责任编辑:JGG
原文链接:https://www.nature.com/articles/s41587-022-01307-0
文章引用:DOI: 10.1038/s41587-022-01307-0








