今天给大家分享一篇2022年6月28日发表在Nature Communications (IF:17.694)上,基于网络的机器学习方法预测癌症患者的免疫治疗反应的文章。

在过去几年中,免疫检查点抑制剂(ICIs)极大地改善了癌症患者的临床治疗。在临床试验中,使用ICIs通常比化疗产生更少的副作用,具有更持久的治疗益处。因此,ICIs已被广泛使用在不同癌症中,包括黑色素瘤、膀胱癌和胃癌。然而,尽管ICI治疗具有临床益处,但是只有少数患者对免疫治疗有反应(实体瘤约30%),ICI治疗后可能出现毒性。因此,需要一种方法来识别能够在给药前检测免疫治疗应答者的生物标记物,提供有关ICIs临床应用的信息,并提高癌症患者的生存率。使用ICIs疗法的一个主要挑战是从免疫疗法治疗的患者中识别标记物,这些标记物可以有力地预测多个癌症患者队列中的药物反应。例如,监测PD1/ PD-L1表达是针对各种癌症类型的伴随诊断试验。因此,许多研究报告了非小细胞肺癌中PD-L1表达与ICI反应之间的正相关。然而,引人注目的是,其他研究报告PD-L1表达与ICI治疗反应之间没有显著相关性,一些研究甚至表明ICI反应者表现出低PD-L1表达水平。这些先前生物标记物的不一致预测,表明急需识别新的生物标记物,以有力地预测免疫治疗反应。Litchfield等人最近发现,传统的生物标记物只能解释约60%的ICI反应,这表明新的因素尚未被发现。由于从接受免疫治疗的患者中识别生物标记物具有强大的挑战,许多最近的研究集中于从未接受ICIs治疗的癌症患者中识别生物标记物。尽管这种方法取得了成功,但这些无监督学习方法的一个主要局限性是,免疫治疗的特异性标记物可能无法从非免疫治疗患者中识别,从而限制了基于ICI的个性化药物的潜在改进。因此,必须开发更精准的方法来识别ICI治疗患者的生物标记物(例如监督学习方法),并最终最大限度地发挥ICI治疗的效益。网络生物学为识别稳健的生物标志物提供了强有力的手段。基于网络的方法利用了具有相似表型作用的基因倾向于共同定位于蛋白质-蛋白质相互作用(PPI)网络的特定区域的观察结果。这种趋势已被用于识别在预测表型结果方面比使用单基因方法更稳健的基因模块。研究表明,在相似网络区域发生体细胞突变的患者表现出相似的临床结果。此外,有文献报道可以从药物靶点和疾病基因之间的接近程度来推断药物的疗效,也可以通过网络邻近性,使用患者衍生类器官模型的药物基因组学数据来识别预测癌症患者总体生存率的药物反应生物标记物。总之,有证据表明,基于网络的方法提供了预测性和低噪声的生物标记物,但该方法的有用性尚未被验证,以预测大样本癌症患者对ICI治疗的反应。研究者建立了一个基于网络的机器学习框架,该框架可以(i)跨ICI数据集进行稳健预测,以及(ii)识别潜在的生物标记物。具体来说,可以使用700多个患者样本中基于网络的生物标记物的表达水平,有力地预测有应答者和无应答者,包括使用针对PD1/PD-L1信号轴的ICIs治疗的黑色素瘤、转移性胃癌和膀胱癌患者。为了识别强大的药物反应生物标记物,实施了一种基于网络的方法,在PPI网络中识别了位于免疫治疗靶点附近的生物学通路。为了衡量该生物标志物的普适性,通过研究交叉验证以及跨研究预测进行了广泛测试。发现,基于NetBio的预测比基于ICI靶点(包括PD1、PD-L1或细胞毒性T淋巴细胞抗原4(CTLA4))的表达水平以及与肿瘤微环境相关的标记物的预测更准确。
之前的工作报道与抗癌药物反应相关的生物标记物位于PPI网络中药物靶点附近。基于之前的工作,研究者通过选择接近ICI靶点的通路来识别与ICI反应相关的生物学通路(图1a,b)。使用了STRING PPI网络(STRING score>700),包括16957个节点和420381条边。首先,应用网络传播(network propagation),使用ICI靶点(例如,nivolumab的PD1或阿替唑珠单抗的PD-L1)作为种子基因,在网络上传播ICI靶点的影响(图1a)。网络传播的一个特点是,距离ICI靶点较近的节点的影响分数较高。接下来,选择了影响分数高的基因(前200个基因),并确定了富含这些基因的生物通路(图1b)。然后,使用选定的生物途径预测免疫治疗反应,并将这些途径视为基于网络的生物标记物(NetBio)。为了进行基于ML的免疫治疗反应预测,使用NetBio作为输入特征;作为阴性对照,使用基于基因的生物标记物(即免疫治疗靶基因)、基于肿瘤微环境的生物标记物或从数据驱动的ML方法中选择的途径(图1c)。利用输入特征的表达水平,应用logistic回归来训练ML模型。为了测试输入特征的预测性能,测量了以下方面的预测性能:(i)通过免疫治疗后缩小的肿瘤大小测量的药物反应,或(ii)患者的生存。在使用监督学习的ML模型中,使用不同的训练和测试数据集组合来广泛测量预测性能的一致性。具体来说,进行了(i)研究内预测,其中训练和测试数据集是从单个队列生成的;或(ii)跨研究预测,其中两个独立的数据集被用作训练和测试数据集(图1d)。此外,交替使用大量或少量的训练样本来衡量各种训练条件下预测性能的一致性。图1:基于网络的机器学习(ML)方法用于识别免疫治疗相关生物标记物2. 研究内交叉验证表明,基于NetBio的ML可以对ICI治疗反应和总体生存率做出一致的预测NetBio标志物可以做出一致的预测性能来预测ICI反应(图2)。相比之下,当使用药物靶点表达时,观察到更强的预测性能。首先使用NetBio或其他已知的免疫治疗相关生物标记物(包括药物靶点)进行了留一交叉验证(LOOCV)来测量性能。为此,使用了四个免疫治疗队列——两个黑色素瘤队列,一个转移性胃癌队列和一个膀胱癌队列。使用NetBio训练的ML模型在所有四个数据集中都做出了准确的预测(图2a-d)。相比之下,使用药物靶点表达水平进行的预测不太一致,其中药物靶点仅在黑色素瘤队列中准确预测(图2a),而在其他三个癌症队列中不准确(图2b-d)。值得注意的是,在Liu数据集中,使用药物靶点的表达水平是反向预测的(图2b)。此外,在三个数据集中,使用基于NetBio的ML预测为ICI反应者的患者的总生存期持续延长;使用药物靶向表达仅在一个数据集中预测总体存活率(图2e-g)。总之,基于网络的方法将生物标记物扩展到药物靶点的网络邻居,改进了基于药物靶点表达水平的预测。
接下来,将NetBio的预测性能与之前确定的其他ICI相关生物标记物进行了比较,发现在大多数情况下,NetBio在所有四个癌症数据集中都更好(图2h-o)。对于单基因标记物,考虑了免疫治疗靶点(PD1、PD-L1或CTLA4)的表达水平。对于肿瘤微环境相关标记物,考虑了与CD8 T细胞比例、T细胞耗竭、CAFs和TAMs相关的基因集。还考虑使用所有单基因标记(GeneBio)或所有肿瘤微环境相关标记(TME-Bio)进行预测。使用准确性和F1分数来衡量LOOCV的预测性能,发现基于NetBio的预测在72个比较中有71个(98.6%)优于使用所有其他生物标志物的预测。这些结果进一步证明,使用基于网络的方法来识别生物标记物可以对癌症患者的ICI反应作出稳健的预测。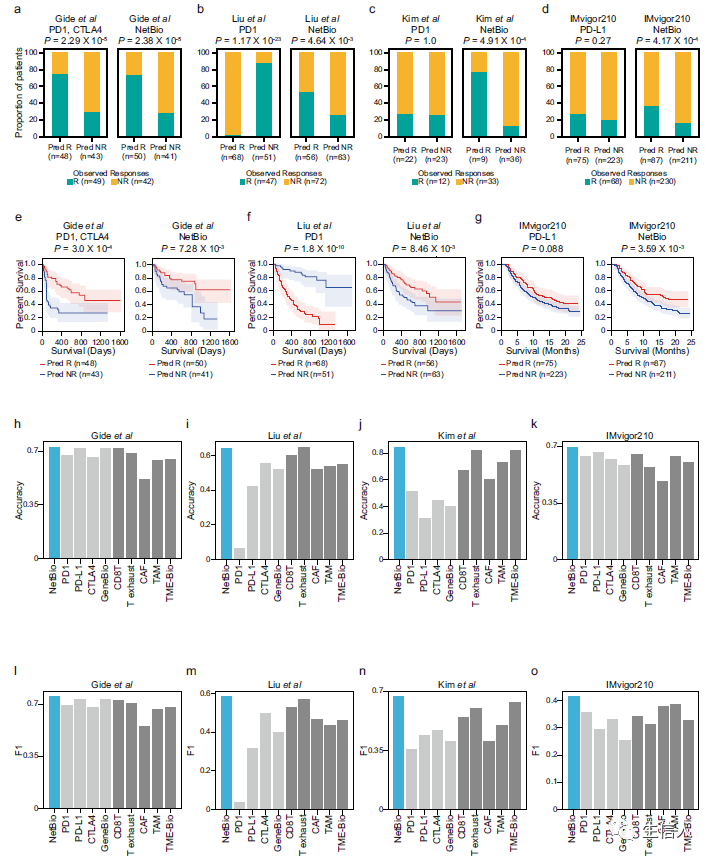 图2: 免疫治疗患者的药物反应和总生存率预测3. 使用基于NetBio的ML的跨研究预测可以在其他独立的黑色素瘤数据集中做出一致的预测精确ML模型的关键方面包括:(i)其推广到新数据集的能力和(ii)在可用训练样本较少时的一致性能。首先,观察到当使用独立数据集时,使用NetBio训练的ML模型可以做出稳健的预测,而当使用其他生物标记物时,预测性能较差(图3)。为了测试ML模型的通用性,使用Gide等人的黑色素瘤数据集来训练ML模型,并在三个独立的黑色素瘤数据集(图3a)中测试预测性能。为了计算该模型的性能,使用了logistic回归模型的预测概率。基于NetBio的ML在两个外部数据集中显示AUC>0.7(图3b,c),在其余数据集中显示AUC>0.69(图3d)。与基于NetBio的ML相比,使用其他生物标记物的预测显示出高度不同的预测性能(图3b-d)。例如,PD-1表达显示出较少的最佳性能,最大AUC仅达到0.66(图3b-d)。此外,尽管在Auslander和Riaz数据集中使用T细胞衰竭标记进行的预测非常准确(图3b,d),但预测性能略优于Prat数据集中的随机预
图2: 免疫治疗患者的药物反应和总生存率预测3. 使用基于NetBio的ML的跨研究预测可以在其他独立的黑色素瘤数据集中做出一致的预测精确ML模型的关键方面包括:(i)其推广到新数据集的能力和(ii)在可用训练样本较少时的一致性能。首先,观察到当使用独立数据集时,使用NetBio训练的ML模型可以做出稳健的预测,而当使用其他生物标记物时,预测性能较差(图3)。为了测试ML模型的通用性,使用Gide等人的黑色素瘤数据集来训练ML模型,并在三个独立的黑色素瘤数据集(图3a)中测试预测性能。为了计算该模型的性能,使用了logistic回归模型的预测概率。基于NetBio的ML在两个外部数据集中显示AUC>0.7(图3b,c),在其余数据集中显示AUC>0.69(图3d)。与基于NetBio的ML相比,使用其他生物标记物的预测显示出高度不同的预测性能(图3b-d)。例如,PD-1表达显示出较少的最佳性能,最大AUC仅达到0.66(图3b-d)。此外,尽管在Auslander和Riaz数据集中使用T细胞衰竭标记进行的预测非常准确(图3b,d),但预测性能略优于Prat数据集中的随机预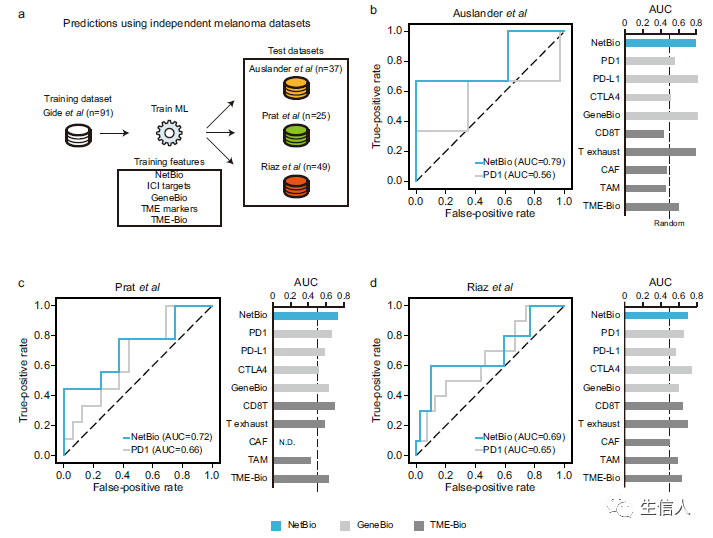 图3: 三个独立黑色素瘤数据集的预测性能4. 基于
NetBio的预测优于纯数据驱动的功能选择方法与纯数据驱动ML预测相比,基于NetBio的ML模型能够持续优化预测性能(图4)。具体来说,对于数据驱动的ML模型,作者选择了在训练数据集中最能区分响应者和非响应者的K个特征(其中K等于NetBio的数量),并使用所选特征训练ML模型(图4a)。在11个不同的任务中,发现基于NetBio的预测比基于ML的特征选择的特征表现出更好的性能(图4b)。 此外,在跨黑色素瘤队列(图4c)进行预测时,一致观察到性能改善,这表明网络传播选择有助于减少ML模型的过度拟合。这一观察结果表明,与纯数据驱动的特征选择相比,网络传播的特征选择可以提供稳健的特征。总之,这些结果进一步表明,可以通过利用基于网络的生物标记物选择来识别稳健的转录组学生物标记物。5. 基于NetBio的预测阐述TCGA数据集中的免疫微环境接下来测试了基于NetBio的预测阐述了TCGA数据集中的免疫微环境特征(图5a)。具体来说,使用Gide或Liu数据集(黑色素瘤队列)在TCGA数据集(TCGA SKCM)中预测黑色素瘤患者的ICI反应,Kim数据集(胃癌队列)预测TCGA胃癌(TCGA STAD),和IMvigor210数据集(膀胱癌队列)预测TCGA膀胱癌(TCGA BLCA)患者,并将预测的药物反应与(i)肿瘤突变负荷(TMB)或(ii)TCGA患者的免疫微环境相关(图5a)。随后,基于NetBio的预测成功地再现了免疫微环境(图5b)。推测Gide和Liu队列的相关结果具有共同特征,因为它们都与黑色素瘤患者有关。正如所料,它们表现出类似的免疫微环境特征,包括与白细胞分数和CD8 T细胞比例高度正相关,与M2巨噬细胞比例高度负相关(图5b)。为了进一步研究了哪个NetBio通路与免疫细胞比例高度相关。使用Gide数据集的ML训练中最重要的通路特征表明,“I类MHC的抗原呈递折叠组装”与CD8T细胞比例呈最高正相关(图5c)。这一发现与预期是相符的,因为抗原呈递细胞或肿瘤细胞的抗原呈递诱导CD8T细胞浸润。使用Liu数据集时,在最重要的通路中,“FGFR信号通路”与CD8T细胞比例的相关性最高,其中通路的表达水平与细胞比例呈负相关(图5d). 此外, 研究者还发现了与胃癌和膀胱癌免疫微环境一致的NetBio通路。在胃癌中,基于NetBio的预测与滤泡辅助性
T细胞比例高度相关(图5b)。在Kim队列中最重要的通路中,“有丝分裂G2-G2-M期”的高表达水平与高滤泡辅助性T细胞比例有关。并且之前的一项研究报道,辅助性T细胞的分化受细胞周期路径的调节。在膀胱癌中,发现基于NetBio的预测与白细胞分数呈正相关(图5b)。以上结果表明,在胃癌和膀胱癌中,也可以通过NetBio途径捕捉免疫微环境。6. NetBio通路的表达水平与膀胱癌患者的免疫细胞浸润有关在膀胱癌患者中,使用其他基于IHC的结果验证了趋化和吞噬通路(即趋化因子受体分别结合趋化因子和FcgR激活)与PD-L1治疗的膀胱癌队列中的免疫浸润相关(图6)。在IMvigor210数据集中使用了不同的免疫表型,包括(i)免疫沙漠(少于10个CD8 T细胞),(ii)排斥(邻近肿瘤细胞的CD8 T细胞)和(iii)浸润(与肿瘤细胞接触的CD8 T细胞)表型(图6a),并将趋化和吞噬通路的表达水平与免疫表型进行比较(图6b、c)。与免疫沙漠或排斥表型相比,免疫浸润表型显示出最高的通路表达水平(图6b,c),表明NetBio通路可以捕捉膀胱癌中的白细胞浸润分数。总之,以上结果表明,NetBio可以很好地揭示与免疫治疗反应相关的免疫微环境相关的通路。图6:NetBio通路的表达水平与膀胱癌中基于免疫组织化学的免疫表型一致7. 将NetBio与ML模型中的肿瘤突变负荷(TMB)相结合,可以优化PDL1抑制剂治疗膀胱癌患者的预测虽然高TMB水平与ICI治疗的益处增加有关,但ICI应答者和无应答者的TMB水平往往存在显著重叠,这表明TMB本身并不是ICI应答的充分预测因子。因此,该工作测试了将NetBio与基于TMB的预测器相结合是否可以提高预测性能(图7a)。将NetBio表达水平与TMB相结合,可以改善使用阿替唑单抗治疗的膀胱癌患者的总体生存率预测(图7b、
c)。使用LOOCV预测ICI治疗反应,仅使用TMB训练ML模型,预测有反应组和预测无反应组之间的1年生存率差异为18%(图7b)。当同时使用TMB和NetBio时,1年生存率差异增加到22.3%(图7c)。在观察到预测性能的改善后,研究者试图确定一个导致预测性能改善的特征,随后发现Raf激活途径在两个亚组之间(R2R vs R2NR)显著差异表达(图7d). 具体来说,根据组合预测模型预测为无应答者的患者(即R2NR患者)显示Raf通路激活。从PPI网络来看,Raf通路的组成部分,包括HRA、KRAS和JAK2和PD-L1直接相邻(图7e),表明该通路可能在药物治疗期间发挥重要作用。为了进一步检验Raf激活通路作为ICI治疗生物标记物的潜在有用性,分析了PD-L1表达、TMB和Raf激活成分的表达水平与整体ICI的相关性体外TCGA膀胱癌数据集的生存率。具体而言,测试了当(i)PD-L1表达较低(模拟PDL1抑制)和(ii)TMB水平较高时,Raf激活是否影响总体存活率。Raf激活通路对表现出低PD-L1表达和高TMB水平的膀胱癌患者的总体生存率有统计学意义的影响(图7f)。重要的是,Raf激活通路的高表达与总体生存率低相关,这一发现与PD-L1抑制剂治疗的患者表现出对治疗的耐药性一致(图7d,f)。总之,以上结果表明:(i)基于网络的转录组生物标记物可以帮助改善基于TMB的免疫治疗反应预测;(ii)可以使用基于网络的方法识别ICI反应生物标记物。图7:结合基于网络的转录组特征和肿瘤突变负荷可以改善PDL1抑制剂(阿替唑单抗)治疗的膀胱癌患者总生存率的预测总之,该工作为使用ICI治疗的精确医学开辟了有趣的新研究机会。例如,开发了一种直接从ICI处理的样本(即监督学习)进行训练的ML方法,而大多数最先进的技术使用从非ICI处理的样本学习的ML模型来预测对ICI处理(即无监督学习)的反应。由于监督和非监督学习使用不同的癌症患者来训练ML模型,因此两种学习方法可以相互补充,在一起使用时可以提高预测性能(例如,半监督方法)。具体而言,当监督学习(NetBio)和无监督学习(Lee等人)的预测彼此之间的相关性较低时,组合预测的性能在所有测试条件下都得到了改善,这表明两种学习方法都可以学习不同但与ICI治疗相关的生物信号。由于免疫治疗的生物学结果非常复杂,依赖单个组学特征的方法在预测患者对免疫治疗的反应方面存在局限性。将基于网络的机器学习模型与不同的组学层相结合将获得更好的临床结果。随着更多的肿瘤样本测序数据可用于ICI治疗和非ICI治疗的癌症患者,我们可以使用机器学习方法做到更精确的预测。
更多生信分析问题咨询:18501230653(微信同号)
图3: 三个独立黑色素瘤数据集的预测性能4. 基于
NetBio的预测优于纯数据驱动的功能选择方法与纯数据驱动ML预测相比,基于NetBio的ML模型能够持续优化预测性能(图4)。具体来说,对于数据驱动的ML模型,作者选择了在训练数据集中最能区分响应者和非响应者的K个特征(其中K等于NetBio的数量),并使用所选特征训练ML模型(图4a)。在11个不同的任务中,发现基于NetBio的预测比基于ML的特征选择的特征表现出更好的性能(图4b)。 此外,在跨黑色素瘤队列(图4c)进行预测时,一致观察到性能改善,这表明网络传播选择有助于减少ML模型的过度拟合。这一观察结果表明,与纯数据驱动的特征选择相比,网络传播的特征选择可以提供稳健的特征。总之,这些结果进一步表明,可以通过利用基于网络的生物标记物选择来识别稳健的转录组学生物标记物。5. 基于NetBio的预测阐述TCGA数据集中的免疫微环境接下来测试了基于NetBio的预测阐述了TCGA数据集中的免疫微环境特征(图5a)。具体来说,使用Gide或Liu数据集(黑色素瘤队列)在TCGA数据集(TCGA SKCM)中预测黑色素瘤患者的ICI反应,Kim数据集(胃癌队列)预测TCGA胃癌(TCGA STAD),和IMvigor210数据集(膀胱癌队列)预测TCGA膀胱癌(TCGA BLCA)患者,并将预测的药物反应与(i)肿瘤突变负荷(TMB)或(ii)TCGA患者的免疫微环境相关(图5a)。随后,基于NetBio的预测成功地再现了免疫微环境(图5b)。推测Gide和Liu队列的相关结果具有共同特征,因为它们都与黑色素瘤患者有关。正如所料,它们表现出类似的免疫微环境特征,包括与白细胞分数和CD8 T细胞比例高度正相关,与M2巨噬细胞比例高度负相关(图5b)。为了进一步研究了哪个NetBio通路与免疫细胞比例高度相关。使用Gide数据集的ML训练中最重要的通路特征表明,“I类MHC的抗原呈递折叠组装”与CD8T细胞比例呈最高正相关(图5c)。这一发现与预期是相符的,因为抗原呈递细胞或肿瘤细胞的抗原呈递诱导CD8T细胞浸润。使用Liu数据集时,在最重要的通路中,“FGFR信号通路”与CD8T细胞比例的相关性最高,其中通路的表达水平与细胞比例呈负相关(图5d). 此外, 研究者还发现了与胃癌和膀胱癌免疫微环境一致的NetBio通路。在胃癌中,基于NetBio的预测与滤泡辅助性
T细胞比例高度相关(图5b)。在Kim队列中最重要的通路中,“有丝分裂G2-G2-M期”的高表达水平与高滤泡辅助性T细胞比例有关。并且之前的一项研究报道,辅助性T细胞的分化受细胞周期路径的调节。在膀胱癌中,发现基于NetBio的预测与白细胞分数呈正相关(图5b)。以上结果表明,在胃癌和膀胱癌中,也可以通过NetBio途径捕捉免疫微环境。6. NetBio通路的表达水平与膀胱癌患者的免疫细胞浸润有关在膀胱癌患者中,使用其他基于IHC的结果验证了趋化和吞噬通路(即趋化因子受体分别结合趋化因子和FcgR激活)与PD-L1治疗的膀胱癌队列中的免疫浸润相关(图6)。在IMvigor210数据集中使用了不同的免疫表型,包括(i)免疫沙漠(少于10个CD8 T细胞),(ii)排斥(邻近肿瘤细胞的CD8 T细胞)和(iii)浸润(与肿瘤细胞接触的CD8 T细胞)表型(图6a),并将趋化和吞噬通路的表达水平与免疫表型进行比较(图6b、c)。与免疫沙漠或排斥表型相比,免疫浸润表型显示出最高的通路表达水平(图6b,c),表明NetBio通路可以捕捉膀胱癌中的白细胞浸润分数。总之,以上结果表明,NetBio可以很好地揭示与免疫治疗反应相关的免疫微环境相关的通路。图6:NetBio通路的表达水平与膀胱癌中基于免疫组织化学的免疫表型一致7. 将NetBio与ML模型中的肿瘤突变负荷(TMB)相结合,可以优化PDL1抑制剂治疗膀胱癌患者的预测虽然高TMB水平与ICI治疗的益处增加有关,但ICI应答者和无应答者的TMB水平往往存在显著重叠,这表明TMB本身并不是ICI应答的充分预测因子。因此,该工作测试了将NetBio与基于TMB的预测器相结合是否可以提高预测性能(图7a)。将NetBio表达水平与TMB相结合,可以改善使用阿替唑单抗治疗的膀胱癌患者的总体生存率预测(图7b、
c)。使用LOOCV预测ICI治疗反应,仅使用TMB训练ML模型,预测有反应组和预测无反应组之间的1年生存率差异为18%(图7b)。当同时使用TMB和NetBio时,1年生存率差异增加到22.3%(图7c)。在观察到预测性能的改善后,研究者试图确定一个导致预测性能改善的特征,随后发现Raf激活途径在两个亚组之间(R2R vs R2NR)显著差异表达(图7d). 具体来说,根据组合预测模型预测为无应答者的患者(即R2NR患者)显示Raf通路激活。从PPI网络来看,Raf通路的组成部分,包括HRA、KRAS和JAK2和PD-L1直接相邻(图7e),表明该通路可能在药物治疗期间发挥重要作用。为了进一步检验Raf激活通路作为ICI治疗生物标记物的潜在有用性,分析了PD-L1表达、TMB和Raf激活成分的表达水平与整体ICI的相关性体外TCGA膀胱癌数据集的生存率。具体而言,测试了当(i)PD-L1表达较低(模拟PDL1抑制)和(ii)TMB水平较高时,Raf激活是否影响总体存活率。Raf激活通路对表现出低PD-L1表达和高TMB水平的膀胱癌患者的总体生存率有统计学意义的影响(图7f)。重要的是,Raf激活通路的高表达与总体生存率低相关,这一发现与PD-L1抑制剂治疗的患者表现出对治疗的耐药性一致(图7d,f)。总之,以上结果表明:(i)基于网络的转录组生物标记物可以帮助改善基于TMB的免疫治疗反应预测;(ii)可以使用基于网络的方法识别ICI反应生物标记物。图7:结合基于网络的转录组特征和肿瘤突变负荷可以改善PDL1抑制剂(阿替唑单抗)治疗的膀胱癌患者总生存率的预测总之,该工作为使用ICI治疗的精确医学开辟了有趣的新研究机会。例如,开发了一种直接从ICI处理的样本(即监督学习)进行训练的ML方法,而大多数最先进的技术使用从非ICI处理的样本学习的ML模型来预测对ICI处理(即无监督学习)的反应。由于监督和非监督学习使用不同的癌症患者来训练ML模型,因此两种学习方法可以相互补充,在一起使用时可以提高预测性能(例如,半监督方法)。具体而言,当监督学习(NetBio)和无监督学习(Lee等人)的预测彼此之间的相关性较低时,组合预测的性能在所有测试条件下都得到了改善,这表明两种学习方法都可以学习不同但与ICI治疗相关的生物信号。由于免疫治疗的生物学结果非常复杂,依赖单个组学特征的方法在预测患者对免疫治疗的反应方面存在局限性。将基于网络的机器学习模型与不同的组学层相结合将获得更好的临床结果。随着更多的肿瘤样本测序数据可用于ICI治疗和非ICI治疗的癌症患者,我们可以使用机器学习方法做到更精确的预测。
更多生信分析问题咨询:18501230653(微信同号)











