今天为大家带来的是于2021年2月发表在Nature Machine Intelligence volume上的一篇文章:A deep learning framework for high-throughput mechanism-driven
phenotype compound screening and its application to COVID-19 drug repurposing,作者从基于表型的高通量数据出发,使用贝叶斯的峰值反卷积技术优化后的LINCS L1000数据集,提出了一种机制驱动的基于神经网络的模型:DeepCE,它利用图神经网络和多头注意力机制来模拟化学结构-基因和基因-基因关联,最终来预测药物作用下的差异基因表达谱。
几十年来,基于靶点的高通量数据筛选占据着药物发现的主导地位,然而,通过此方法得到的药物最终只有极少部分能最终批准成为权威的治疗药物。基于表型的高通量数据筛选比基于靶点的药物发现具有很多优势,但存在吞吐量低、靶向去卷积困难等问题,因此,本文的研究面向高通量、机制驱动的表型化合物筛选方法。本文从基于表型的高通量数据筛选出发,使用贝叶斯的峰值反卷积技术优化后的LINCS L1000数据集,提出了一种机制驱动的基于神经网络的模型:DeepCE,它利用图神经网络和多头注意力机制来模拟化学子结构-基因和基因-基因关联,最终来预测全新化学物质扰动下的差异基因表达谱。此外,本文提出了一种新的数据增强方法,该方法从LINCS L1000数据集中的不可靠数据中提取有用信息。实验结果表明,DeepCE的性能较好。在下游分类任务和COVID-19的药物再利用中表现同样较好。本文用到的数据集有如下几个:
LINCS L1000:LINCS L1000数据集包含不同基因在不同剂量、不同药物、不同细胞下的表达谱,本文使用该数据集训练、验证DeepCE和其他模型。原始的L1000质量较低,有一些技术可以提高此数据集的质量,如高斯混合,本文使用的是基于贝叶斯的峰值反卷积技术处理后,根据L1000中Level 4的数据筛选出的高质量Level 5数据集,筛选方法为计算二者之间的平均Pearson相关系数(APC),选择APC分数大于等于0.7的样本作为高质量数据。最后本文将高质量数据集分成了训练集、验证集和测试集,各数据集详情如表1所示。其中,augmented 的数据集将在下文描述。
表1.L1000数据集详情 STRING:STRING是一个多元的蛋白质-蛋白质相互作用数据库,这个作用可以是已知的或预测的、直接的或间接的。蛋白质间的作用来源于基因组环境预测、高通量实验、之前的知识数据库等多个地方,共包含约19000个蛋白质和约1200万条相互作用信息。本文使用该数据库通过node2vec方法计算L1000数据集中基因的向量标表示,此外,本文同样使用该数据库来计算药物靶点的信息。DrugBank:DrugBank是包含药物及其靶点信息的在线数据库,本文使用该数据库提取多种药物及其属性。
STRING:STRING是一个多元的蛋白质-蛋白质相互作用数据库,这个作用可以是已知的或预测的、直接的或间接的。蛋白质间的作用来源于基因组环境预测、高通量实验、之前的知识数据库等多个地方,共包含约19000个蛋白质和约1200万条相互作用信息。本文使用该数据库通过node2vec方法计算L1000数据集中基因的向量标表示,此外,本文同样使用该数据库来计算药物靶点的信息。DrugBank:DrugBank是包含药物及其靶点信息的在线数据库,本文使用该数据库提取多种药物及其属性。COVID-19病人基因表达谱(NGDC, NCBI),本文使用该数据来计算患者和正常人之间的差异基因表达谱,之后与药物作用下的基因表达谱进行比较,以此来寻找可能治愈COVID-19的药物。
各数据在本文工作流程中的作用可概括为图1。
本文建立的模型由多个模块组成,首先,作者使用GCN网络提取药物的特征,并使用前馈神经网络量化细胞系和药物剂量特征,同时,作者基于STRING数据库基于Node2Vec方法将基因数据编码为向量表示。然后将这些向量输入到使用了注意力机制的相互作用网络中,以此来捕获药物-基因、基因-基因间的关联。最后,将各模块输出的向量输入到预测模块中,预测所有基因的表达值。模型的总体结构见图2。
在许多生物信息预测问题上使用数据驱动的药物化学指纹比预定义的药物化学指纹(例如PubChem、ECFP)更有效,因此,本文使用GCN来捕获化学亚结构信息。
在基于神经网络的模型中,一个集合的元素基于注意力权重选择性地将注意力聚焦于另一个集合(attention)或者它本身的集合(self-attention)的注意力机制被广泛应用于人工智能应用中,尤其是在计算机视觉和自然语言处理中表现非常好。本文使用一种称为多头注意力的注意力机制来提取基因和药物结构之间的关联性特征。
这种多头注意力机制是构建DeepCE相互作用模块的主要部分。DeepCE的相互作用模块由两个相同的层组成,其中第一层的输出作为第二层的输入,对于每一层,作者使用了两个独立的多头注意力模块,每个模块有4个头。query,key 和value向量的长度为512。注意力模块的输出经过连接、归一化之后流入到一个前馈层和归一化层中。
预测层接受连接到一起的药物化学特征、基因特征、细胞系特征和药物剂量特征,通过具有ReLU激活函数的线性层,最终输出978个基因的表达值,978及为L1000数据集中的基因数量。
作者使用均方误差(MSE)作为训练DeepCE模型的目标函数。
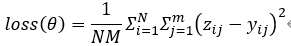
其中,θ是DeepCE模型的参数集合,N和M分别是数据集中基因表达谱的个数和L1000中基因的个数。zij和yij分别是在第i个基因表达谱中第j个基因表达值的真实值和预测值。
作者在实验中使用的几种基线模型分别是线性模型、Vanilla神经网络、KNN和TT-WOPT。
作者测试了一个多输出的线性回归模型及其正则化版本的模型,包括Lasso回归(L1正则化)模型和岭回归(L2正则化)模型。于DeepCE类似,线性模型输入药物、基因、细胞系和药物剂量的数值表示,有一点不同的是在线性模型上输入的是预定义的药物化学特征,而不是数据驱动的通过GCN模型得到的化学特征。多输出线性模型可以看作是一层具有激活函数的前馈神经网络。
本文使用的vanilla 神经网络有点像DeepCE的简化版本,它不包括基因-基因、基因-药物之间的相互作用模块和生成药物结构特征的GCN模型。该模型的输入类似于线性模型,模型中的层类似于DeepCE中的预测模块,它包含两个拥有ReLU激活函数的前馈层。
本文还提出了一种基于kNN的方法用来预测全新药物作用下的基因表达。具体来说,在特定条件下(细胞系、药物剂量)的全新药物作用下的基因表达由与它条件相似的训练集中邻近样本的平均基因表达值决定。作者实验了从1到15不同近邻样本数量下的模型表现,以及在不同距离度量方法下的模型表现,比如余弦相似度、Jaccard、Tanimoto和欧几里得距离。
TT-WOPT全称为Tensor-train weight optimization,是一种张量补全优化技术,用于从已有的张量数据中补全缺失值,该方法在预测L1000数据集中缺失值的有效性已经被证实。由于该模型不需要额外的信息,因此该模型的输入是作为张量表示的L1000基因表达值。
从图3中可以看出只有小部分的样本是可靠的(APC分数大于等于0.7),如果我们不能从不可靠的样本中提取可靠信息的话那将会浪费大量数据。在表2中可以看到,如果直接将不可靠的数据加入到高质量数据集中(原始数据集),将会使模型的表现变差,因此,本文提出了一种数据增强方法,通过这种方法可以有效地利用不可靠的实验样本来提高模型的性能。
作者认为,虽然有一些样本(Level 5数据)是不可靠的,但不一定其对应的Level 4数据也不可靠,作者提出的数据增强方法尝试找出这些Level 4可靠的数据。该方法的基本思想是首先在高质量数据集上训练模型,然后为这些不可靠的数据由模型生成对应的基因表达谱,并于其原来的基因表达谱进行比较,若二者的相似性大于某个阈值,则将该样本纳入数据增强后的数据集中,作者在衡量相似性的方式上选用的是Pearson相关系数。
在衡量模型表现性能方面作者主要用到了Pearson相关系数,它比传统的MSE更能反应真实值和预测值之间的相似性。除了Pearson相关系数,作者还使用了其他衡量方式,有RMSE、GSEA、Positive@k。其中,Person相关系数和RMSE关注所有的基因表达,而GSEA和Positive@k只关注部分最重要基因。使用多种度量方式可以从不同方面衡量模型的准确性。
作者对结果的描述主要基于Person相关系数,同时兼顾其他指标。
作者将DeepCE及其几个变体与几个基线模型进行了比较,包括vanilla神经网络、kNN、线性模型和TT-WOPT。DeepCE的几个变体分别为DeepCE 对于DeepCE,作者使用神经指纹(neural fingerprints,及前述通过GCN得出的药物特征)作为药物特征的描述方法,而对于其他模型,作者使用的是预定义的化学指纹,包括PubChem、ECFP6指纹和药物靶点信息。所有模型都在高质量数据集上训练、验证。
如表2所示,DeepCE及其变体的性能比基准模型有了极大的提升。特别是DeepCE模型在测试集上获得了0.4907的Person相关性,大大超过了其他模型。与去除了相互作用模块的简单变体相比,DeepCE也取得了更好的性能,表证明了对化学结构-基因和基因-基因之间特征的关联性进行建模的有效性。在基准模型中,vanilla神经网络和kNN取得了较好的分数,而线性回归、Lasso回归、TT-WOPT和岭回归表现极差,基本没什么作用。这表明线性关系不足以对此数据集中变量之间的依赖关系进行建模。
作者为了深入研究模型的预测性能,探索了测试集和训练集之间药物化学相似性的影响。作者首先选择测试中的一个样本然后计算训练集中同细胞系下与之药物最相似的样本之间的差异,这种差异由两种药物PubChem指纹的TAnimoto系数确定。在计算了测试集中所有样本到训练集的差异后,作者按升序对它们进行排序,然后将数据均匀分成4份。从图4中可以看到所有模型都是在药物差异更低的数据集上表现会更好,另外DeepCE在同差异性时于vanilla网络和kNN模型表现更好。
表2中已经可以大概看出模型的性能受数据集质量影响,本小节作者基于vanilla神经网络和kNN算法更加细致地探讨了数据集质量对模型性能的影响,结果如图5所示。在APC值小于0.1时,模型在预测时的结果基本没有可靠性,当APC值大于0.1时模型渐渐起作用,并随着APC值的升高模型的性能也在不断升高。这些结果表明不可靠的数据对模型预测的性能有严重的负面影响,从数据集中去除这部分数据是实现良好预测性能的必要条件。
为了进一步证明DeepCE的价值,作者尝试了使用药物影响下的基因表达谱来寻找治疗新冠肺炎的药物。首先使用在高质量数据集上训练好的DeepCE模型来生成DrugBank数据库中所有11179种药物在最大剂量作用下预测的基因表达谱。对于新冠肺炎患者的基因表达谱,作者使用来自NGDC和NCBI的SARS-CoV-2基因表达数据集来计算患者的基因表达谱,输入的细胞系为A549——一种肺癌细胞系。然后根据Spearman相关系数选择患者的基因表达数据与得分最低预测的基因表达数据对应的药物作为新冠肺炎的潜在药物。表3显示了模型预测的最终结果,在这前10种药物中,所有的药物至少都已经有人注意到了该药物对新冠肺炎的有效性,其中的药物有已经被用在了丙型肝炎中的、有已经成为蛋白酶抑制剂的、还有作为抗生素和治疗肺炎的药物。这表明了DeepCE在预测新冠肺炎潜在药物中的有效性。
本文从基于表型的高通量数据筛选出发,使用贝叶斯的峰值反卷积技术优化后的LINCS L1000数据集,提出了一种机制驱动的基于神经网络的模型:DeepCE,它利用图神经网络和多头注意力机制来模拟化学子结构-基因和基因-基因关联,最终来预测新型药物作用下的基因表达谱。
此外本文提出了一种新的数据增强方法,该方法从 L1000 数据集中的不可靠数据中提取出了有用信息。
实验结果表明,DeepCE模型的预测性能较好。在COVID-19的药物再利用中具有较高的有效性。








