
大规模并行 DNA 测序导致生物学中高度多重实验的快速增长。这些实验产生独特的测序结果,需要特定的分析管道来解码高度结构化的读数。然而,尚未开发出解释测序读数以提取其编码信息用于下游生物分析的多功能框架。
在这里,不列颠哥伦比亚大学和东京大学的研究人员报告了 INTERSTELLAR(interpretation, scalable transformation, and emulation of large-scale sequencing reads,大规模测序读数的解释、可扩展转换和仿真);理论上,它可以解码任何类型的测序读数中编码的数据值,并将它们转化为另一种选择结构的测序读数。
INTERSTELLAR 成功地从一系列短读长和长读长测序读数中提取了信息,并翻译了单细胞 (sc)RNA-seq、scATAC-seq 和空间转录组学的那些数据,从而方便研究人员用不同软件工具进行分析。INTERSTELLAR 将极大地促进基于测序的实验的开发和数据分析管道的共享。
该研究以「A universal sequencing read interpreter」为题,于 2023 年 1 月 4 日发布在《Science Advances》。

在过去的几十年里,利用微阵列和高通量 DNA 测序,DNA 条形码的概念使一系列汇集的生物筛选成为可能。早期的例子包括建立酵母缺失集合,其中每个菌株都被构建为在缺失位点具有两个独特的 DNA 条形码。可以汇集条形码酵母菌株并进行单一生长竞争测定,其个体相对生长变化可以通过竞争前后微阵列或高通量测序测量的条形码数量读出。该策略开创了化学基因组学领域筛选药物靶基因的先河。
不久之后,同样的概念也被应用于基于哺乳动物细胞培养的全基因组基因敲除和敲除分析。在这些测定中,细胞由编码短发夹 (sh) RNA 或 CRISPR-Cas9 引导 (g) RNA 的慢病毒文库转导。由不同扰动引起的细胞生长可以通过聚合酶链反应 (PCR) 扩增和小 shRNA 或 gRNA 编码 DNA 片段的测序来大量量化。
此外,产生远端基因组区域和与不同因素相关的 DNA 条形码的嵌合融合的实验系统,使得研究人员能够大规模探索染色质构象、蛋白质相互作用、遗传相互作用和单分子 RNA 的空间细胞分布。在单细胞和空间基因组学中,单细胞标识符 (ID)、空间 ID 和唯一分子 ID (UMI) 用于唯一标记相应的转录组或基因组 DNA 片段,这导致了单细胞 RNA 测序(scRNA-seq)、scATAC-seq、空间转录组学和空间基因组技术的发展。
上述方法中的每一种都可以同时进行多个实验并生成测序文库。来自不同检测的测序文库也可以通过将额外的文库特异性、独特的 DNA 条形码融合到每个测序文库 DNA 中,进一步复用用于单次测序运行。这些实验的输出 DNA 分子具有一系列复杂性,其中一些编码多个信息片段,其组合有时被设计为通过多个读取(例如,配对末端读取和索引读取)读取。
然而,存在一些共同的问题——这些基于测序的实验中的大多数方法,都是使用它们自己专有的软件工具针对特定的序列读取结构开发的。虽然许多此类工具具有先进的下游数据分析功能,但它们通常不能重复用于概念上相同类型的实验系统产生的测序读数。对于具有改进的性能和不同读取结构的概念相同的分析,已经反复提出新的实验方法,并且已经为它们各自的读取结构开发了处理基本相同信息的数据分析工具。
在 scRNA-seq 领域尤其观察到这些轮子的再发明。这些软件工具不能交换不同的 scRNA-seq 库结构,也不能通过将它们应用于相同的 scRNA-seq 数据集来进行交叉验证。已经做出多项努力来开发能够分析特定类别实验的不同读取结构的灵活软件工具,例如 UMI-tools、zUMIs、scumi(用于基于 UMI 的 RNA-seq 和 scRNA-seq)和 SnapATAC(对于 scATAC-seq),但它们对于正在进行的产生独特读取结构的新实验的开发无效。
任何测序数据分析都遵循每次读取中序列片段的识别(例如,在 scRNA-seq reads 中识别细胞 ID、UMI 和 cDNA 编码区域)以及提取的序列片段和值(例如,映射到参考基因组和 scRNA-seq 中每个 RNA 种类的 UMI 计数)的下游分析。
因此,不列颠哥伦比亚大学和东京大学的研究人员提出了两种解决方案:(i) sequencing read interpreter 和数据分析工具的开发——如果一个 read interpreter 只提取在 sequencing reads 中编码的数据值,那么它的数据分析 pipeline 应该适用于产生相同数据结构的其他实验的 sequencing reads;(ii) read translator 的开发——如果可以将某种格式的测序 reads 翻译成另一种 reads 结构,则可以使用为特定 reads 结构开发的现有数据分析管道来分析其他 reads 结构。基于这两个方案,研究人员开发了称为 INTERSTELLAR 的单一通用工具。
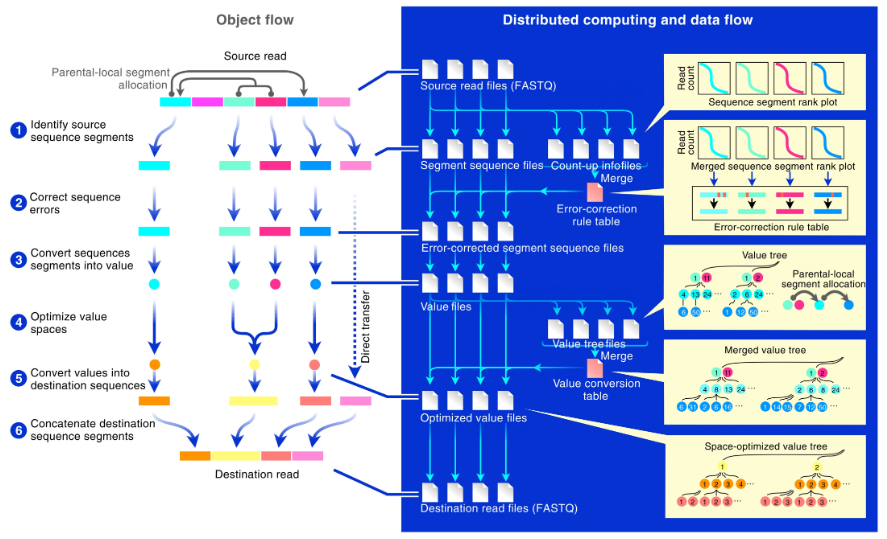
图示:INTERSTELLAR 的概述。(来源:论文)任何测序文库的结构都是通过用序列片段的位置规定在DNA序列中编码的信息或使用恒定标记序列对其进行切片来设计的(否则测序后无法分析文库)。在对文库进行测序之后进行任何测定后,提取序列片段并进行错误校正以用于下游分析。INTERSTELLAR 完全有能力使用灵活的正则表达式系统和序列段中编码的值的亲本关联来解码任何这些读取。
研究人员使用不同的软件工具对 scATAC-seq、scRNA-seq 和空间转录组学读数进行读数翻译和数据分析,并将结果与原始专有软件工具分析的原始读数进行比较。尽管原始结果和仿真结果的总体结果非常相似,但观察到的差异程度不同。
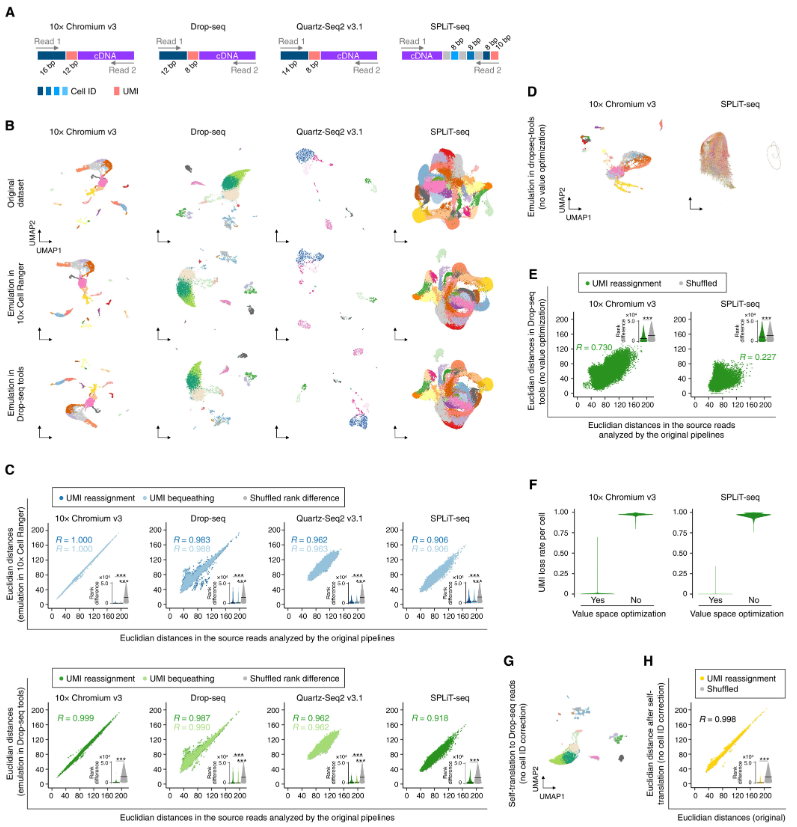
图示:不同 scRNA-seq 读数和软件工具的交叉评估。(来源:论文)
结果的差异可以通过三个潜在来源来解释:(i) 读取解释过程,(ii) 目标段分配过程,以及 (iii) 不同软件工具之间价值分析过程的差异,其中 INTERSTELLAR 负责前两个。从 scRNA-seq 读取翻译演示中,读取解释过程的纠错步骤被认为是所见差异的潜在主要来源,其中读取解释的纠错很可能使在不同软件工具中实施的纠错步骤无效(即,通过 INTERSTELLAR 覆盖纠错策略)。

图示:多模式 scRNA-seq 读取的翻译。(来源:论文)
虽然 Levenshtein 距离度量是 INTERSTELLAR 的基于非许可名单的纠错的默认值,并且对于大多数测序读取数据分析来说这实际上不是问题,但它可以用 Bartender 或用户开发的插件代替。当目标片段的信息容量(或代表性)小于相应源片段的信息容量(或代表性)时,目标片段序列分配过程是源读取中编码信息丢失的唯一潜在来源。
为了解决这个问题,研究人员在理论上实施了最佳价值空间优化策略,该策略使用亲本段分配的用户定义信息,并成功地证明了读取翻译的信息损失可以最小化,同时降低了序列代表性。
在过去的几十年里,除了临床样本和各种物种的(表观)基因组学和转录组学分析之外,大规模并行短读长测序技术的应用使得广泛的生物检测得以发展,并且该领域继续迅速扩大。虽然开发专有测序读数解释器和数据分析管道并将其与新的基于测序的分析方法的开发结合起来是一种实践,但该团队的研究人员建议应开发下一种形式,即社区可使用通用的测序读数解释和翻译平台, 如 INTERSTELLAR,只开发数据分析部分,单独共享,以最大限度地利用数据处理资源。
论文链接:https://www.science.org/doi/10.1126/sciadv.add2793
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。









