论文题目:
Direct generation of protein conformational ensembles via machine learning
今天给大家介绍Giacomo Janson等人在Nature Communication上发表的文章“Direct generation of protein conformational ensembles via machine learning”。动力学和构象取样是将蛋白质结构与生物功能联系起来所必需的。虽然在实验上很难探测,但计算机模拟被广泛用于描述蛋白质动力学,然而计算成本很高,限制了可以研究的系统。在这里,本文证明了机器学习可以用模拟数据进行训练,直接生成物理上仿真的蛋白质构象集合,而不需要任何采样,并且计算成本可以忽略不计。作为原理证明,本文训练了一个基于transformer架构的生成对抗网络,该网络具有对本质无序肽的粗粒度模拟的自注意力机制。由此产生的模型idpGAN可以预测训练集中不存在的序列的依赖于序列的粗粒度集成,表明可以在有限的训练数据之外实现可移植性。本文还在原子模拟数据上重新训练idpGAN,以表明该方法在原则上可以扩展到更高分辨率的构象集成生成。

蛋白质的生物学功能不仅仅由其单一的三维结构决定,还取决于其动态特性,从而形成构象集合。表征构象集合对于机械地理解蛋白质的活性和调控至关重要,并对生物医学科学、生物技术和药物设计产生影响。实验技术在研究生物分子动力学时存在空间或时间分辨率低的限制。因此,计算方法被应用于研究蛋白质动态并生成结构集合。物理学基础的分子动力学模拟是一种强有力的策略。分子动力学模拟可以从分子系统的可能构型中采样,以识别构象空间中能量最有利的区域。然而,高维度和重要的动力学壁垒导致除了最简单的蛋白质系统外,即使使用专门的计算机硬件或增强采样方法,也会产生极大的计算挑战。因此,需要新的策略来加速生成具有生物学意义的动态集合。
近年来,基于数据驱动的机器学习技术成功地解决了给定氨基酸序列预测蛋白质三维构象的挑战。机器学习方法(如AlphaFold2)在准确性方面已经达到了实验确定的结构的水平。同时,这些方法面临了一些挑战,主要包括:(1)蛋白质是动态实体,具有多种构象状态,如何对构象集进行建模,从而提高预测准确性;(2)如何针对给定系统生成之前未见过的分子构象,并为具有不同化学组成的新系统生成构象;(3)如何设计高效的条件生成模型来生成蛋白质构象集。解决这些挑战将有助于提高蛋白质构象预测的准确性和计算效率,并推动蛋白质相关领域的发展。本文使用基于分子力学模拟数据训练的条件生成模型来生成蛋白质构象。这项研究选择无序蛋白质作为研究对象,因为这种蛋白质具有构象可变性。研究人员使用生成对抗网络(GAN)来学习构象数据集中的三维构象分布,并使用所提出的模型idpGAN来输出三维笛卡尔坐标,并能够对具有不同序列和长度的构象进行建模。该模型具有快速的抽样能力,可以在短时间内生成数千个独立的构象,提供了计算高效的方式来生成构象集。
本文主要提出了一个基于transformer架构的生成对抗模型idpGAN。
idpGAN是一种用于生成无序蛋白质(IDP)构象集合的条件生成模型。该模型的主要特点是可以在给定蛋白质序列的情况下,直接生成符合蛋白质性质和约束的构象集合。该模型的结构主要由生成器网络和判别器网络两部分组成。生成器网络是用于生成符合特定蛋白质序列的构象集合的核心组成部分。在idpGAN中,研究人员使用了一种名为Transformer GAN的变体GAN,该GAN的生成器网络基于transformer架构。具体地,生成器网络包含了一组transformer编码器和解码器,其中编码器将输入序列转换为一组隐藏状态,解码器则将隐藏状态转换为输出序列。这些隐藏状态可以被用于自适应地学习不同输入序列之间的关系,从而提高了生成器网络的性能。此外,生成器网络还包含了一个前馈神经网络,用于将生成器网络输出的Cα坐标转换为三维笛卡尔坐标。判别器网络是用于判断生成的构象集合是否符合蛋白质的性质和约束的部分。在idpGAN中,判别器网络包含了一组残差网络,其中每个残差块包含了两个卷积层和一个残差连接。这些卷积层用于提取构象集合的特征表示,而残差连接则用于将原始输入与残差块的输出相加,从而减少模型训练过程中的梯度消失问题。在训练过程中,idpGAN使用了一种条件变量,即蛋白质序列,来指导生成器网络生成符合特定蛋白质序列的构象集合。此外,研究人员还使用了一种变分自编码器(VAE)来提高模型的泛化能力。具体地,VAE可以将输入数据压缩到一个潜在空间中,并将这个潜在空间作为条件变量输入到生成器网络中,从而提高模型对未知数据的泛化能力在训练idpGAN时,研究人员使用了多种计算模拟数据,如残基级别的粗粒化模拟、全原子隐式溶剂模拟和全原子显式溶剂模拟等,从而提高模型的泛化能力。在模型训练完成后,研究人员对模型进行了多个实验数据集的测试和验证,结果表明,idpGAN能够生成具有实验数据一致性的蛋白质构象集合,并且比其他传统的分子模拟方法更快速和高效。需要注意的是,idpGAN生成的蛋白质构象集合是基于Cα表示的,而不是全原子表示。Cα表示是一种简化的蛋白质表示方法,它只考虑了蛋白质的主链,而忽略了侧链的影响。因此,idpGAN生成的构象集合可能会受到这种简化表示方法的限制。不过,这种简化表示方法也提高了模型的计算效率,使得idpGAN能够在较短的时间内生成大量的独立构象。
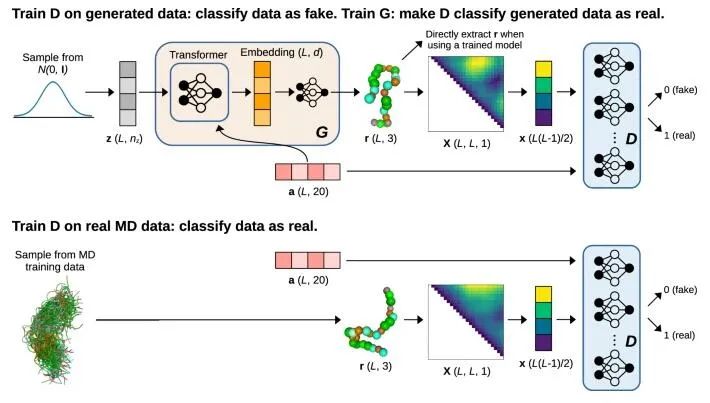
图1 IdpGAN网络架构图
基于粗粒化模拟的idpGAN
作者使用由残基蛋白模型获得的MD数据训练idpGAN,训练后,在CG MD数据上对31个选定的idp进行了模型评估,命名为IDP_test,这些idp不在训练集中,并且在其中没有类似的序列。同时,还将长度为20至200的聚丙氨酸(polyAla)链作为无序列特异性相互作用的随机线性聚合物,与CG MD模拟的数据进行了比较,以研究idpGAN是否可以提供更好的近似。下图显示了来自IDP_test set的his5、protac和htau23k17蛋白的MD (reference) (a)、idpGAN (b)和polyAla (c)集合的平均力分布图。对于每个蛋白质,从集合中得到的构象沿着参考MD数据拟合的PCA模型的前两个分量进行投影。idpGAN生成的图再现了非常相似的独特模式,即使protac(或类似的IDP)不存在于训练集中。这清楚地表明,idpGAN从训练MD数据中学习了可转移的残基特异性相互作用模式。最后,图2还显示了idpgan生成的模型的旋转半径和能量分布(基于CG模型能量函数)与md生成的系综非常吻合。这表明该模型在估计旋转半径分布方面具有实用价值,并且它们具有高结构质量,没有冲突或其他明显违反立体化学约束。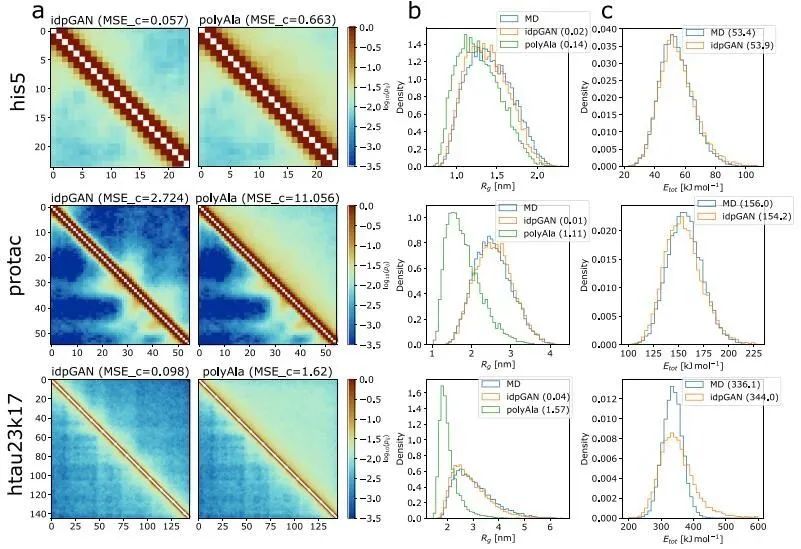
图2 idpGAN中三个IDP_test蛋白的例子基于主成分分析(PCA)的平均力势进行了分析,以进一步表征由idpGAN产生的集成。首先使用原子间距离作为输入特征对参考MD数据进行PCA。然后将idpGAN的构象投影到由MD数据定义的PCA空间上。为了控制,还从具有相同数量氨基酸的聚ala链的MD模拟中投影快照。在图3中,作者绘制了三个选定的idp_test蛋白质沿前两个主成分的自由能图。发现idpGAN提供了很好的近似MD集成,而polyAla集成显示出明显的差异,特别是对于protac和htau23k17。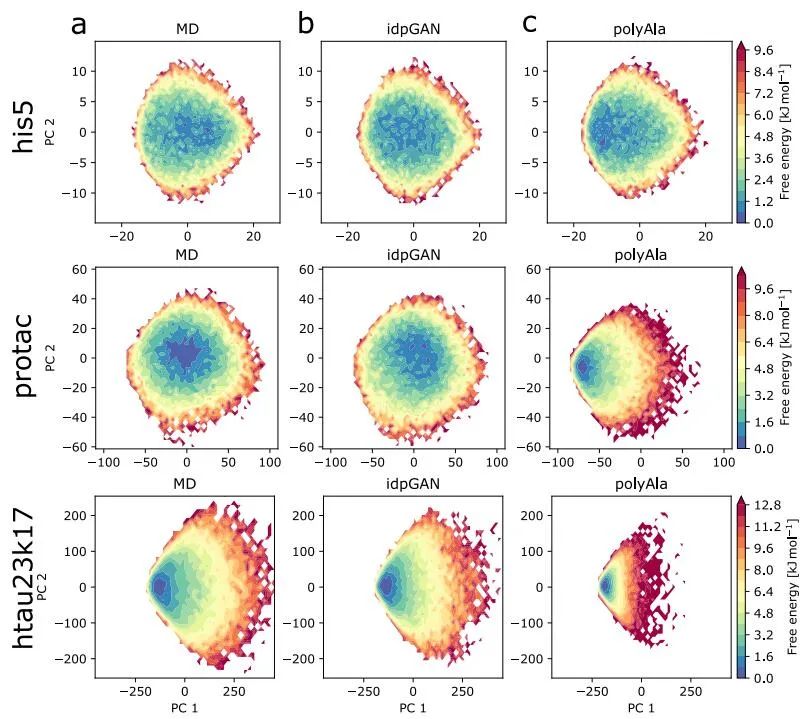
在所有IDP_test蛋白中评估不同的指标,以定量评估idpGAN(图4)。接近零的值反映了MD参考集合的更好近似值。同样的指标也计算了相应的polyAla集合,以提供一个随机的聚合物基线。另一个基线是通过从另一个独立的长期MD运行中绘制快照来计算的,并将这些快照与用于评估idpGAN的相同参考模拟的MD快照进行比较。该基线本质上捕获了同一系统不同MD模拟轨迹之间采样的可变性。这些结果表明idpGAN产生能量稳定的构象,以可转移的方式捕获了MD数据中集成的可变性及其氨基酸序列特异性特征。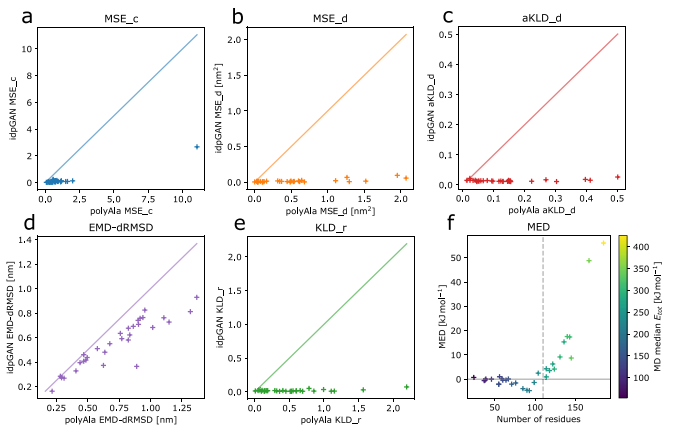
图4 评估idpGAN和polyAla集成近似MD数据
基于全原子隐式溶剂模拟的 idpGAN
用全原子隐式溶剂马尔可夫链蒙特卡罗(MCMC)模拟的数据重新训练idpGAN,使用ABSINTH势,可以准确地概括实验确定的几种idps的性质。ABS_test肽(P27205, P02338.0, Q9EP54和Q2KXY0)的合成示例如图5所示。对于大多数肽,idpGAN正确地捕获了参考MCMC集成的相关特征。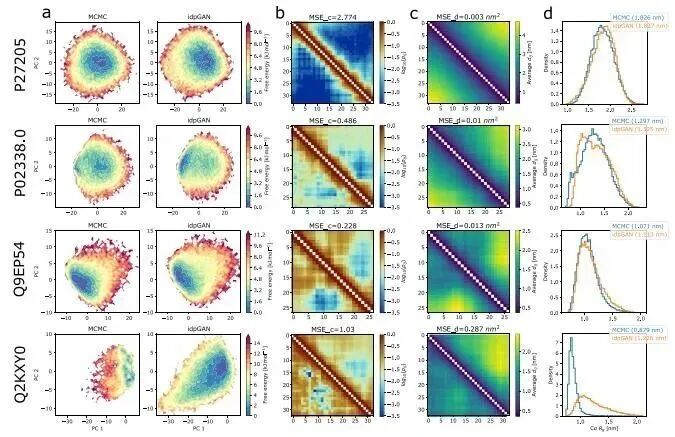
图5 来自idpGAN的四个ABS_test肽的例子基于全原子显式溶剂轨迹的 idpGAN
在这个实验中,作者模拟了大IDP的显式溶剂MD轨迹的Cα痕迹。实验结果表明,像idpGAN这样的网络有潜力模拟更复杂的全原子蛋白质构象,这是通过显式溶剂模拟获得的。作者推测,如果类似idpGAN模型能够成功完成这一无条件任务,那么如果使用来自不同蛋白质的足够的轨迹数据进行训练,它应该能够推广到其他序列。idpGAN采样速度
为了比较CG MD模拟和idpGAN发生器的采样速度,作者测量了两者用于生成足够样本的GPU时间,以恢复在5 μs MD运行中观察到的旋转半径分布。图7a显示了idpGAN和MD集合的KLD_r,与长MD集合相比,包含越来越多的样品。对于G网络和MD模拟,随着更多的构象被采样,KLD_r得到改善,并最终在MD中达到零。对于his5、protac和htau23k17, G网络在KLD_r(称为tgen)中达到平台所需的计算时间总是小于3秒。MD模拟达到相同的KLD_r值(称为tMD)所需的时间总是在250秒以上。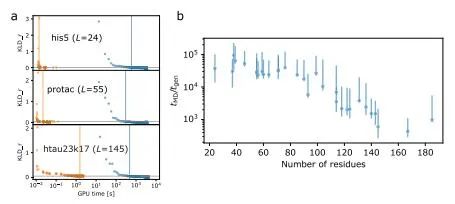
图6 与MD相比,评估IDPGAN采样效率
本篇论文的研究背景是蛋白质构象预测,旨在解决现有蛋白质构象预测方法不能准确反映蛋白质构象动态性的问题。为此,研究人员提出了一种名为idpGAN的生成对抗网络模型,通过对分子动力学模拟数据进行训练,可以直接生成蛋白质构象集合,从而提高预测的准确性和多样性。该模型基于transformer架构,能够在保证高效性和可扩展性的同时生成高质量、多样性的蛋白质构象集合。在论文中,研究人员通过使用粗粒化模拟、全原子隐式溶剂模拟和全原子显式溶剂轨迹数据,对idpGAN进行了全面的评估。结果显示,idpGAN生成的构象集合与实验测定的构象集合之间具有非常高的相似性和多样性。同时在运行速度方面,也具有较快的效率。
https://www.nature.com/articles/s41467-023-36443-x









