将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑 | 绿萝
化学反应的预测建模在药物发现和材料科学等各个行业中起着至关重要的作用。然而,由于化学转化的复杂性和多样性,实现具有所需外推能力和化学可解释性的合成转化预测模型具有挑战性。
为了弥补丰富的化学领域知识与先进的分子图模型之间的差距,来自浙江大学洪鑫课题组提出了一种嵌入数字化空间和电子信息的基于知识的分子图模型:SEMG-MIGNN。此外,还开发了分子相互作用模块,以了解反应组分的协同影响。
该研究证明了这种基于知识的图模型可以很好地预测反应产率和空间选择性,其外推能力得到了实验证实。由于局部环境的嵌入,该模型可以在原子水平上解释空间和电子对整体合成性能的影响,这为分子工程实现目标合成功能提供了有用的指导。该模型为反应性能预测提供了一种外推和可解释的方法,指出了化学知识约束反应建模对于合成目的的重要性。
该研究以「Reaction performance prediction with an extrapolative and interpretable graph model based on chemical knowledge」为题,于 2023 年 6 月 15 日发布在《Nature Communications》上。

SPR 预测
对化学的理解和对反应性和选择性的准确预测,为合理、高效地探索大规模合成空间提供了基础。结构-性能关系(Structure–performance Relationship,SPR)的建立主要集中在反应机理的研究和决定性过渡态模型的阐明。
利用过渡态模型,化学家可以阐明观察到的反应性/选择性趋势的来源,并根据化学理论和经验进行合成判断。但在没有明确的机制基础和解析方程的情况下处理高维 SPR 是一个挑战。
数据驱动的方法最近已成为 SPR 建立的强大策略。通过利用合成数据中的相互关系,现代机器学习(ML)算法可以为合成预测创建强大的模型。然而,人工合成转化的机器学习预测和设计还远未成熟。主要瓶颈之一是适合 SPR 预测的分子编码方法和 ML 框架的可用性。
除了人类指定的分子编码工程之外,化学预测领域对表征学习的兴趣也越来越大。通过表征学习的创新和应用,数据驱动的分子性质和反应性能预测取得了重大进展。
特别是在 SPR 预测方面,MIT 的 Coley 及其同事将经典的图神经网络(GNN)模型与选定的反应位点量子化学描述符相结合,开发了一种名为 QM-GNN 的融合模型。这种融合模型将特定于位点的电子信息嵌入到 ML 建模中,从而提高了对一系列转换的区域选择性和反应性的预测能力。QM-GNN 模型的成功表明,增强局部化学信息的表达可以为约束合成建模提供有价值的支持。
受 QM-GNN 模型的启发,研究人员推测可以通过丰富化学环境的局部编码和加强反应组分之间的信息相互作用来进一步改进 SPR 预测。
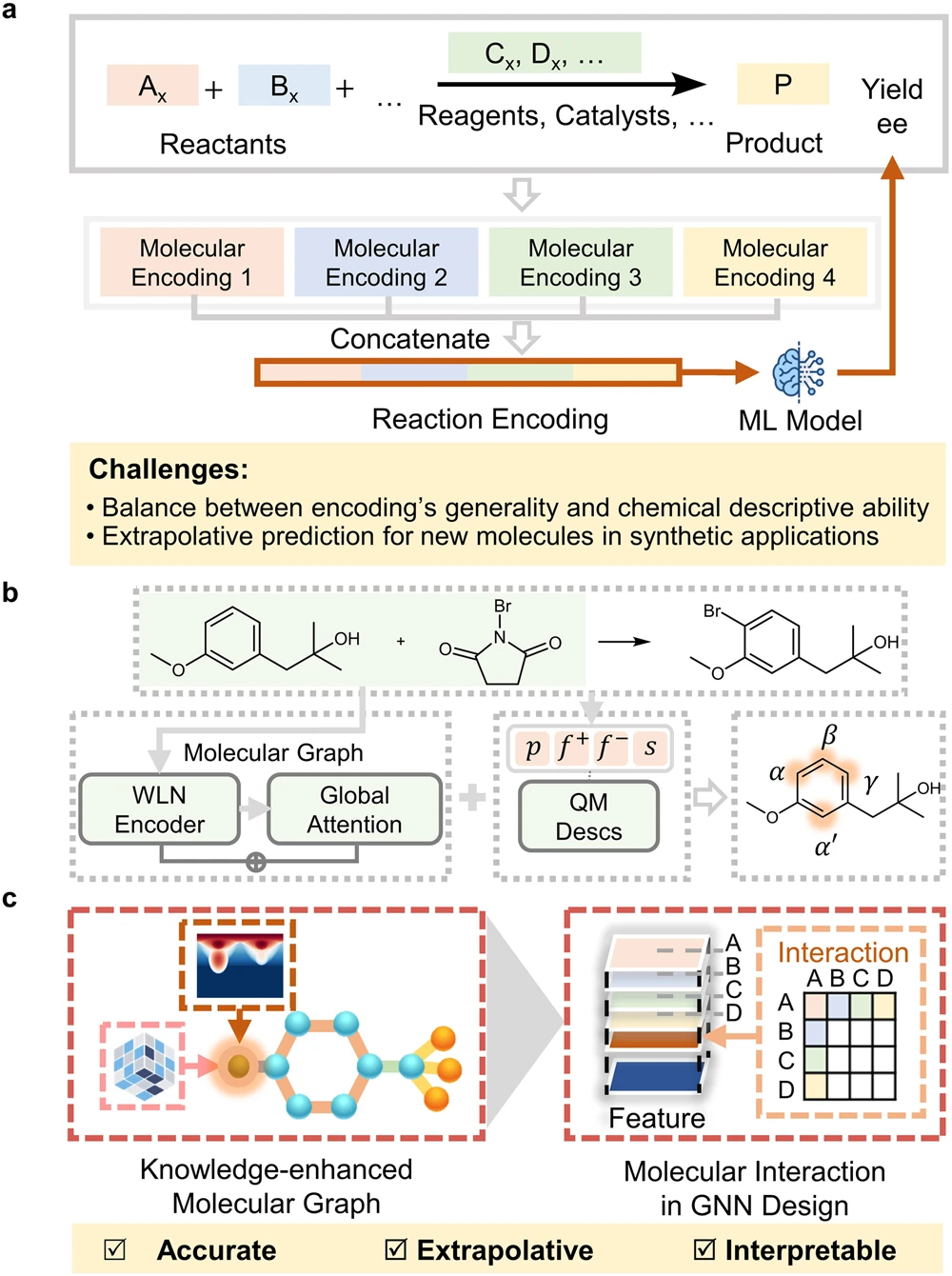
图 1:合成性能和分子特性的机器学习预测。(来源:论文)基于化学知识的 ML 模型用于预测合成性能
在此,研究人员开发了一种名为「基于分子相互作用图神经网络的空间和电子嵌入分子图」(Sterics-and Electronics-embedded Molecular Graph with Molecular Interaction Graph Neural Network,SEMG-MIGNN)的基于化学知识的 ML 模型,用于预测合成性能。SEMG-MIGNN 有两个主要创新设计:首先,空间和电子效应的局部化学环境被数字化并嵌入到图表示中。这显著丰富了模型对原子环境的表征,并提高了模型对新分子结构的外推能力。此外,分子相互作用模块可以有效地学习多个反应组分的协同控制,从而使分子建模有效地扩展到 SPR 建模领域。
具体而言,该研究引入了一种称为空间和电子嵌入分子图(SEMG)的分子图模型,该模型结合了外部化学知识,以提高模型对局部化学环境的区分能力。该知识增强分子图的设计的关键是在节点中嵌入空间和电子环境的原子信息。图 2 以 1-氯-4-(三氟甲基)苯为例,展示了设计的分子图模型的生成工作流程。
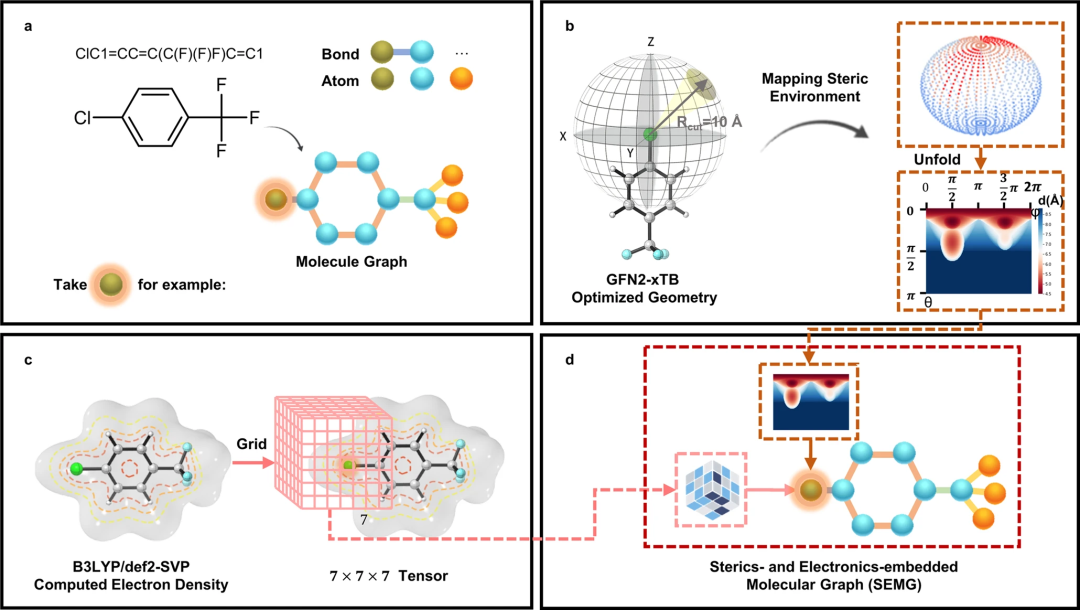
基于 SEMG 丰富的化学信息,接下来,研究人员修改了 GNN 的框架,使其适用于 SPR 预测。特别是,开发了分子相互作用模块以增强模型训练过程中反应组分之间的信息交换。与超图不同,研究人员设计的分子相互作用 GNN(MIGNN)依赖于矩阵运算来实现信息交换。
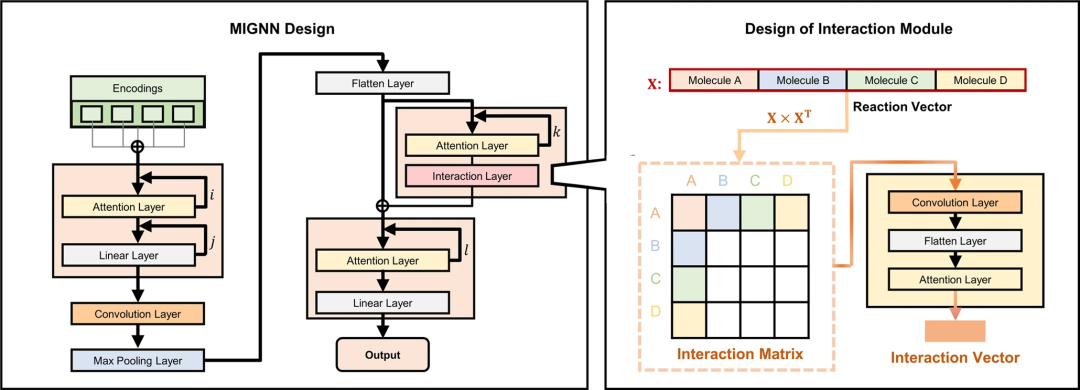
图 3:分子相互作用图神经网络(MIGNN)的框架和相互作用模块的设计。(来源:论文)MIGNN 的交互模块为 ML 模型提供了捕捉反应组分协同效应的机会,这对传统的 SPR 预测 ML 框架具有挑战性。在 MIGNN 中,相互作用矩阵允许具有物理意义的反应组分的所有可能组合充分交换它们的编码信息,从而支持模型在反应组分交织的高维合成空间中的预测。
模型性能预测
该模型在一系列具有挑战性的任务中获得了出色的产率和空间选择性预测。
在 Pd 催化的 C-N 交叉偶联反应的产率预测和手性磷酸(CPA)催化的硫醇加成 N-酰基亚胺的对映选择性预测中发现了很好的预测。对额外的 CPA 催化剂的进一步实验测试证实了该模型在新催化剂预测中的外推能力。特别是,研究发现 SEMG-MIGNN 模型在基于支架的拆分任务中表现出卓越的外推能力,考虑到需要扩展可用 SPR 数据的领域,这对于合成预测来说是非常可取的。
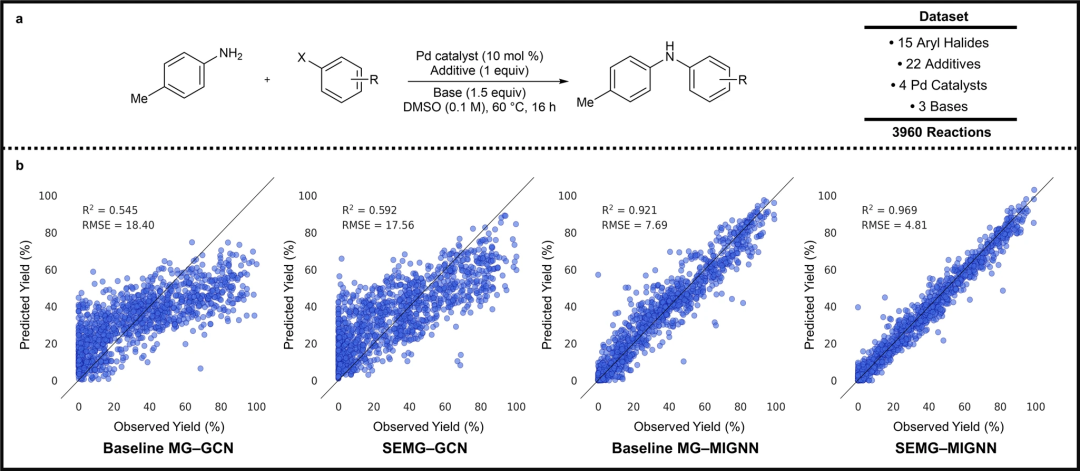
图 4:SEMG-MIGNN 模型(空间和电子嵌入分子图与分子相互作用图神经网络)预测反应产率。(来源:论文)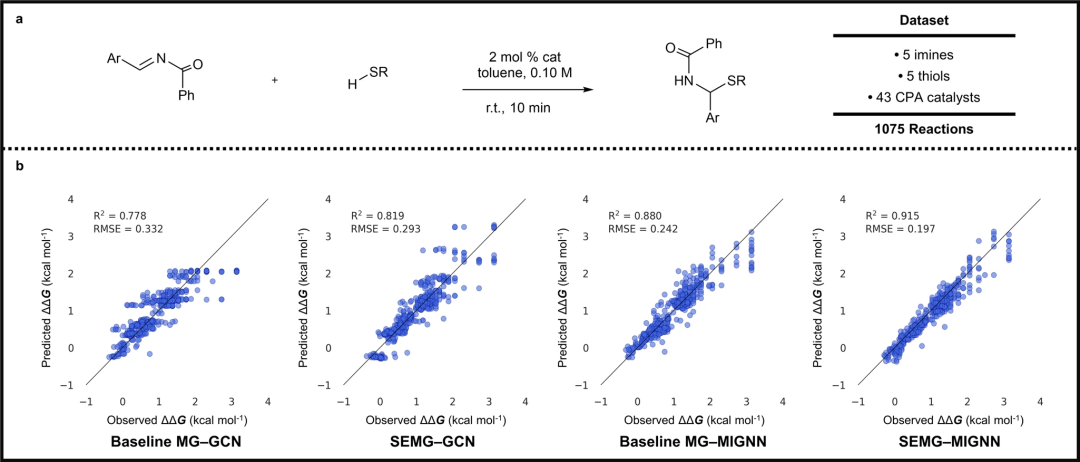
图 5:SEMG-MIGNN 模型预测对映体选择性。(来源:论文)除了出色的预测能力外,空间和电子效应的物理意义编码还提供了原子级的化学解释。对训练模型的分析表明,电子效应对 C-N 交叉偶联的产率预测起着至关重要的作用,而对映体选择性预测则严重依赖于空间效应。
此外,该模型能够识别分子结构的热点,用于确定合成性能,为未来的设计提供有用的见解。该模型的有效性表明,将表示学习与数字化化学知识相结合可以支持化学空间中可泛化模型的开发,为数据驱动的合成转换设计提供机会。
论文链接:https://www.nature.com/articles/s41467-023-39283-x
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看
。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。











