今天为大家介绍的是来自Nature Communication的一篇关于通过深度学习方法来评估RNA-蛋白质复合物类原生结构的文章。本文提出一种新的RNA-蛋白质复合物评分函数——DRPScore,该方法明确考虑结构灵活性。使用基于物理的模拟来生成训练诱饵,用于训练评分函数,并在RNA-蛋白质测试集上对DRPScore进行广泛评估。

如今已经开发了几种计算方法来评估RNA-蛋白质结构,这些方法可以分为基于倾向的统计评分函数和原子级的统计评分函数。基于倾向的评分函数统计分析了成对核苷酸-残基的接口倾向,基于玻尔兹曼逆公式构建了势统计公式。例如,DARS-RNP24是Tuszynska和Bujnicki引入的一种粗粒度的基于倾向的评分函数,DARS-RNP24通过四个项构建了评分函数:空间冲突惩罚和对距离、角度和位置的依赖性。随后,Xiao等人以核苷酸-残基对的构象为统计变量,基于统计势能构建了一种新的评分函数3dRPC-Score,它使用相对RMSD(均方根偏差)来评估核苷酸-残基对之间的构象差异来反映能量。基于倾向的评分函数可以考虑基于成对的核苷酸-残基相互作用,但考虑构象变化具有挑战性。
原子级统计评分函数服从玻尔兹曼分布的距离依赖相互作用势,在类原生结构评估中比基于倾向的评分函数更具判别性。例如,ITScore-PR23是黄仁勋和邹某开发的一种原子级统计评分函数,其核心思想是通过比较训练集中预测原子对和原生原子对之间的差异,通过迭代来提高原子间对的电位。ITScore-PR23在绑定对接方面非常有效,但对未绑定对接具有挑战性。
本文中提出了一种基于深度学习的RNA-蛋白质复合物评分函数,以明确考虑结构灵活性。作者使用基于物理的模拟来生成训练诱饵,用于训练基于深度学习的评分函数。然后,在RNA-蛋白质测试集上对DRPScore进行广泛评估,包括unbound状态的RNA-蛋白质。结果显示出明显改进,并且在选择类原生RNA-蛋白质复合物方面DRPScore始终优于现有方法。
传统RNA-蛋白质复合物评分函数:统计势函数假设不同原生结构特征的分布服从玻尔兹曼分布。然后,这些方法计算了界面相互作用构建能量函数的概率,并识别出能量最低的类原生复杂结构。
DRPScore评分函数:DRPScore没有使用整个RNA-蛋白质结构,而是专注于6 Å距离内的RNA-蛋白质相互作用接口。作者提取了截止值为6 Å的RNA-蛋白质接口结构。其次,利用 RNA 核苷酸中具有质量和电荷的 85 种原子类型和蛋白质残基中具有质量和电荷的 225 种原子类型来考虑原子水平的相互作用。然后,将相互作用接口信息与网格中原子的占领数、质量和电荷的累积一起输入卷积神经网络。在每个核苷酸和残基上创建一个 32 Å 网格,其局部笛卡尔坐标由原子指定。

3D卷积神经网络方法:3D 方法独立处理每个核苷酸以生成局部表示,然后在最后应用平均池化以生成序列的全局表示。每个核苷酸seqi由
3D卷积模块独立处理:OL=Conv3D(OL-1),其中OL-1是形如CL-1×HL-1×WL-1×DL-1的张量,表示第L-1层的RNA核苷酸。Conv3D将通道数由CL-1投影到CL并下采样空间维度。最后,通过平均每个核苷酸的单个表示来生成 RNA 序列的全局表示,但这样可能会丢失有关核苷酸相互作用的关键信息:

4D卷积神经网络方法(DRPScore):作者提出的4D方法通过在序列维度上加入额外的卷积运算来解决3D
CNN方法的缺点。也就是说,4D CNN方法不仅捕获了空间信息,还捕获了序列信息(即核苷酸/残基之间的相互作用)。作者的Conv4D方法使用大小为3的核苷酸/残基的非重叠移动窗口来捕获卷积每一层核苷酸/残基之间的相互作用,通过使用多层CNN可以捕获更远的核苷酸/残基之间的相互作用。
网络共有有六层,最后一层是用于分类的全连接层。前六层中的每一层都有一个Conv4D模块、一个BatchNorm模块(可选)和一个MaxPooling模块。Conv4D模块中的通道数分别为[64,128,256,512,512],步长分别是[2,2,2,1,1]。所有池化模块都使用8的内核大小和192的步长,最后一个池化是全局平均池化,它将空间维度减少到1。获得RNA-蛋白质复合物的最终整体表示为

本研究中的 4DCNN 捕获了蛋白质和 RNA 的序列、二级结构特征和三级结构特征。重原子的序列、质量和电荷被认为是 4DCNN 中的局部特征。另一方面,二级结构(蛋白质的α螺旋和β片、茎、假结、内环、发夹环、单链和RNA连接)以及每个核苷酸和残基之间的距离被认为是全局特征。此外,还充分提取了RNA-蛋白质结合接口的相互作用,包括静电相互作用、范德华相互作用、氢结合和π-π堆积相互作用。
训练集:从NDB数据库中提取了951个RNA-蛋白质复合物结构。经过去除了长度为10个核苷酸的短RNA;考虑了ITScore-PR23中描述的不超过6条蛋白质或RNA链的病例;用cd - hit以0.95序列相似性截断去除了RASP47和DRNA11 RNA
冗余。从346个无冗余的结构中随机选取了277个进行训练,剩余的结构被进一步处理构建bound-bound测试集。使用3dRPC生成RNA-蛋白质结构的诱饵,为训练集中的每一个复合物生成10000个诱饵,根据RMSD选取排名前500的诱饵,即每个复合物有一个原生结构和500个对接结构。
测试集:作者用两个独立的RNA-蛋白质对接测试集测试了DRPScore。非冗余的bound-bound RNA-蛋白质对接基准。从剩余的非冗余RNA-蛋白质复合物中随机选择了36个,生成了三个bound-bound RNA-蛋白集,用3dRPC为这三组中的每个复合物生成1000个诱饵。非冗余的unbound RNA-蛋白质对接基准(由Huang和Zou提供)。该基准保留了57个RNA-蛋白质unbound复合物结构,其中有41个unbound-unbound结构合16个unbound-bound结构,用3dRPC为每个复合物生成1000个诱饵。
预测评估标准:RNA-蛋白质复合物预测的质量通过CAPRI标准进行评估。Irmsd指的是相应蛋白质叠加后的天然结构和预测结构之间的接口RMSD。其中RMSD的定义是
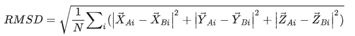
如果预测的复合物和天然复合物之间的小于等于4.0
Å,定义为成功预测。
在bound/unbound RNA-蛋白质测试集上的测试:在三个生成的bound RNA-蛋白质检测集上评估DRPScore。DRPScore的性能始终优于ITScore-PR、DARS-RNP24和3dRPC-Score。结果表明,在前80(20)个预测中,DRPScore表现更好,平均成功率为56.79%(91.67%),而ITScore-PR为63.70%(89.81%),DARS-RNP为37.64%(86.11%),3dRPC为3.5%(33.3%)。

图2 DRPScore和其他评分函数在bound测试集上的性能进一步关注相对困难的
unbound案例,以评估DRPScore在具有挑战性的案例中是否始终表现更好。在前43(20)个预测中,DRPScore的成功率为86.5%(56.14%),而 ITScore-PR为38.60%(45.61%),DARS-RNP为35.09%(40.35%),3dRPC 为 84.3%(11.3%)。总体而言,DRPScore 的性能明显优于ITScore-PR、DARS-RNP和3dRPC。

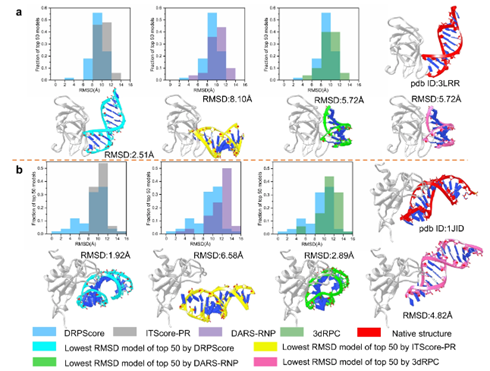
图3 DRPScore和其他评分函数在unbound测试集上的性能作者为前5、10和20个模型中的每个评分函数选择了得分最高的模型。对于每种RNA-蛋白质复合物,记录了整个模型中最低的RMSD。当接口处相互作用核苷酸/残基的数量相对较少时,DRPScore在RNA-蛋白质模型上表现最佳,而其他评分函数对交互作用变化过于敏感,导致结果不尽如人意。

图4 在unbound-bound测试集上类原生结构排名的详细分析为了进一步评估不良采样结构的性能,作者分析了与dsRNA(PDB ID:3LRR)结合的human RIG-I CTD的前3个得分最高的结构模型,作为一个bound实例。DRPScore的最低RMSD模型为2.51 Å,而ITScore-PR为8.10 Å,DARS-RNP为5.72 Å,3dRPC为72.3 Å。DRPScore的平均RMSD为8.94 Å,优于其他三个评分函数中约10.0 Å的RMSD。总之,DRPScore导致预测的分布向较低的RMSDs转移。
基于物理的相互作用贡献:现有的RNA-蛋白质复合物评价方法都是基于玻尔兹曼公式的统计势函数。DRPScore使用深度学习来评估,可以从训练集中的各个帧中准确地提取更多特征。例如,使用HBPULS计算枯草芽孢杆菌YxiN蛋白与23 S核糖体RNA片段(PDB ID:3MOJ)接口的氢键,DRPScore鉴定的氢键中有62.5%与实验结构一致。ITScore-PR、DARS-RNP 和3dRPC均未鉴定出任何天然氢键。
比传统深度学习模型的进步:传统的3DCNN提供并学习局部结构特征,但不考虑全局结构特征。DRPScore模型学习到核苷酸内(局部)和核苷酸-残基间(全局)信息。这是通过在序列维度上添加卷积层来实现的,每一层都逐渐模拟核苷酸-残基之间更长范围的相互作用。故DRPScore模型可以提供、学习局部和全局结构特征,包括二级结构相互作用。
总之,作者开发了一种有效的评分函数,使用基于深度学习的方法评估RNA-蛋白质复合物。DRPScore在不同的测试集上识别RNA-蛋白质复合物的类原生结构的能力已经过广泛的评估。与其他可用方法相比,如果考虑前80个(56)个预测,DRPScore的绑定对接成功率高达91.67%(43.86%),unbound对接的成功率高达56.14%(5.20%)。这些显著的改进表明,DRPScore解决了RNA-蛋白质复合物结构评估的关键灵活性问题。作者期望该方法有助于RNA相关预测和药物开发。










