1.残基共同进化与蛋白质功能适应性相关联
蛋白质序列中的突变可能以非独立的方式影响适应性,这也被称为遗传相互作用或表观相互作用。研究发现,通过对蛋白质进行深度突变扫描(DMS)量化的表观相互作用可以用来推断蛋白质的接触和结构。由于结构上接近的蛋白质残基通常是通过序列进化中的共变对推断出来的,研究人员假设共进化信息也可用于推断蛋白质的表观性或适合度。
为了验证这一假设,研究人员研究了残基对的共同进化与双突变体的适应性之间的关系。收集了一项对人类YAP65 WW结构域双突变体的适应性进行测量的DMS研究,还通过将直接耦合分析模型拟合到WW结构域的同源序列来量化残基间的成对依赖关系的强度。
研究发现,成对依赖关系的强度与双突变体的适应性呈正相关(Spearman相关系数为0.35;图1a)。研究人员还使用依赖关系强度的变化(通过将突变序列与野生型序列进行对比)来预测一系列DMS研究中蛋白质变体的适应性。研究发现,预测与实验数据的相关性在0.1至0.5之间(图1b)。此外,研究人员观察到,如果一个蛋白质具有更多的同源序列,相关性得分会呈上升趋势,这可能是因为丰富的同源序列会导致直接耦合分析模型拟合得更准确。总的来说,这些结果表明可以利用进化信息来预测蛋白质适应性。这促使研究人员将蛋白质序列的进化信息整合到一个监督模型中,以增强对定向进化中蛋白质变体适应性的预测能力。
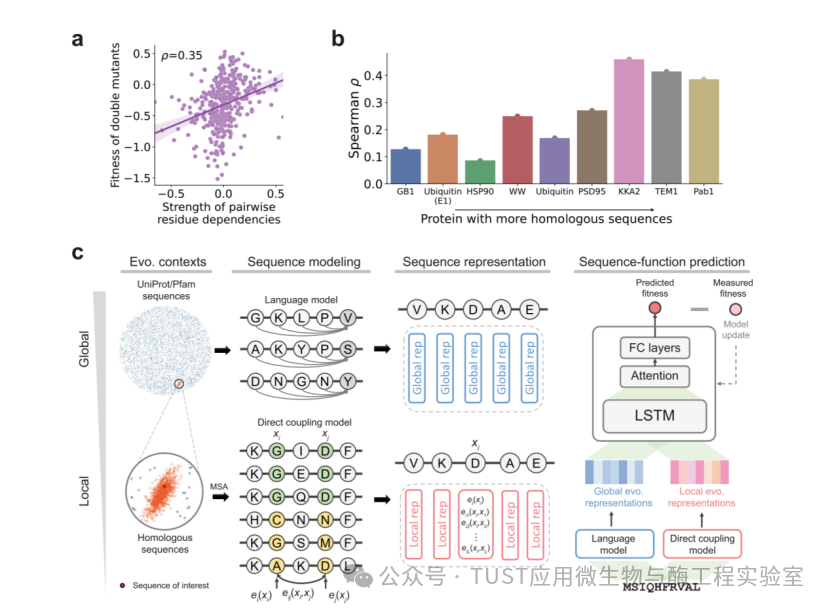
图1 进化上下文整合的蛋白质工程序列建模方法的动机和概述
2.序列到功能建模
研究人员建立了一个深度学习序列到功能模型ECNet,它能从数据(如深度突变扫描测得的适配性)中学习从蛋白质序列到各自功能的映射(图1c)。研究人员使用 LSTM 神经网络架构,并利用大规模深度突变扫描数据集训练蛋白质特异性模型。
该模型主要得益于两种信息丰富的蛋白质表征,一种表征特定蛋白质的残基相互依存关系,另一种表征蛋白质宇宙中的一般序列语义。现有工具(如PolyPhen-2和CADD)通过独立考虑每个氨基酸来预测突变的保护效应,而其他工具(如FoldX和OSPREY)则利用结构信息。然而,蛋白质的功能往往是由蛋白质中残基之间的相互依赖关系(如外显关系)驱动的,而且并非所有的蛋白质结构都已被解析。因此,研究人员通过从同源序列或序列族中提取进化保守信号,对蛋白质中所有位点对的相互作用进行明确建模。利用同源序列的多序列比对(MSA)拟合生成图形模型,以揭示定义同源序列家族的潜在约束或相互依存关系。这些制约因素是自然选择下进化过程的结果,可能揭示出哪些突变比其他突变更可容忍或更有利的线索。
除了相关蛋白质的特定进化序列上下文外,全局蛋白质序列上下文(即编码结构和稳定性的序列上下文)也可以为该预测模型提供信息,以预测突变的影响。为此,研究人员整合了无监督蛋白质上下文语言模型中的一般蛋白质序列表征。语言模型通过使用UniProt和Pfam等大型蛋白质序列语料库,以特定氨基酸周围的所有其他氨基酸为语境,学习预测该氨基酸出现在某一位置的可能性。在训练过程中,语言模型会逐渐改变其内部动态(编码为隐藏状态向量),以最大限度地提高预测准确性。研究发现,通过使用语言模型的隐藏状态向量作为输入特征来微调特定任务的监督模型,可以改进与蛋白质相关的各种科学任务,包括二级结构预测、接触预测和远程同源性检测。在这里,研究人员还将语言模型的隐藏状态向量作为预测模型的另一种蛋白质序列表征,以捕捉全局蛋白质序列上下文(图1c),这是对局部进化上下文表征的补充。
局部和全局进化表征共同用于为感兴趣的蛋白质序列建模。然后,深度学习模型(递归神经网络)将这些序列表征作为输入,学习序列与功能之间的关系。定量功能测量(如通过深度突变扫描测量的适配性数据)用于监督深度学习模型的训练。
3.准确预测蛋白质的功能适应度
为了验证 ECNet,研究人员进行了多项基准实验,以评估 ECNet 预测蛋白质序列功能适配性的能力。
首先将进化上下文表示法与蛋白质序列或突变的不同表示方案进行了比较。Yang等人建议使用在约50万个UniProt序列上预先训练的Doc2Vec模型将任意序列映射到64维实值向量。为了直接测试序列表示法的实用性,研究人员将深度学习模型用作本研究的表示法和Yang等人的Doc2Vec表示法的预测器。在Envision数据集上比较了这两种方法,该数据集由12项DMS研究组成,生成了10种蛋白质的单个氨基酸变体的适合度值。研究发现,在所有12个数据集上,ECNet的表现始终优于Yang等人的方法,就实现的Spearman相关性而言ECNet的相对改进幅度从16%到60%不等(图2a)。
由于Doc2Vec表征是从UniProt数据集中学习的,因此它捕获的信息主要是蛋白质的一般属性,而不是序列中决定功能的依赖关系。相比之下,本研究的进化上下文表示法明确地模拟了序列中残基对的外显性,它们以非独立的方式共同影响着功能。这种细粒度信息使预测模型能够更有效地学习序列-功能映射,从而提高预测性能。研究人员将此进化上下文表示法与Envision模型进行了比较,后者使用27种生物、结构和理化特征来描述单个氨基酸的替换。与这种方法相比,ECNet在不使用这些特征的情况下仍然提高了大多数蛋白质的Spearman相关性。
由于蛋白质工程的重点是识别比野生型具有更好特性的变体,研究使用分类指标(AUROC分数)进一步评估了模型的性能,将功能测量值高于野生型序列的变体定义为阳性样本,其余变体定义为阴性样本。观察到11/12个蛋白质DMS数据集的AUROC分数都有类似的提高(图2b)。这些结果表明,序列上下文比变异氨基酸的描述符信息量更大,这对于捕捉残基之间的相互依赖关系以预测功能至关重要。
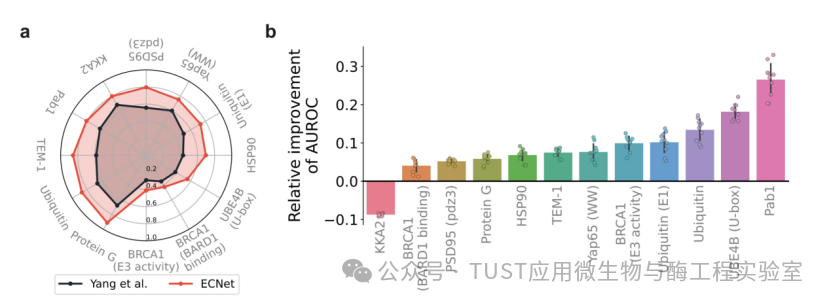
图2 与其他蛋白质变体表示方法的比较
研究人员将ECNet与其他序列建模方法进行了比较,以便在之前策划的一组更大的DMS数据集上预测突变效应。首先将ECNet与三种无监督方法进行了比较,包括EVmutation、DeepSequence和Autoregressive。这些方法在同源序列上训练生成模型,并通过计算突变序列和野生型序列的序列概率对数比来预测突变效应。与 EVmutation、DeepSequence和Autoregressive相比,使用监督预测器预测突变效应的 ECNet 在几乎所有蛋白质上的表现都优于这些方法(图3a)。
研究人员还将ECNet与两种监督方法进行了比较。一种是TAPE,它使用蛋白质语言模型学习到的序列表示作为输入,训练一个与ECNet具有相同模型架构的神经网络。另一种是UniRep,它使用自己的语言模型的输出来训练神经网络。研究发现,ECNet结合了全局LM表征、局部进化表征和原始序列作为输入,在几乎所有蛋白质上都比单独使用LM表征的TAPE和 UniRep获得了更高的相关性(图3b)。
研究人员还进行了消融分析,以剖析模型输入中每个表征成分的性能,结果发现使用联合表征的模型优于使用任何单独表征的模型。此外,还模拟了在有噪声的训练数据上训练ECNet并在无噪声数据上进行测试的实验。结果表明,ECNet 对数据噪声具有鲁棒性。总之,在大量DMS数据集上进行测试后,ECNet的表现明显优于其他序列建模方法,无论是无监督还是有监督的(图3c),这表明它在预测蛋白质变体的适应性景观方面能力出众。

图3 与其他突变效应预测序列建模方法的比较
除了评估 ECNet 在预测所有变体适应性方面的性能外,研究人员还进一步设计了一项实验,以评估ECNet优先处理高性能变体的能力。为此训练了一个ECNet模型,将其用于预测随机拆分的测试集中的所有变体,并根据其预测适配度对其进行排序。然后,计算了真正的前100个变体中被排在ECNet预测的前K个变体的比例。
该实验模拟了定向进化的过程,即在K个变体的测序预算下,识别并合成最有潜力的变体进行筛选。在avGFP、GB1和Pab1三个DMS数据集上,ECNet的召回率更高,而且与UniRep和EVmutation相比,ECNet能更有效地发现适应度最高的变体。与随机抽样方法相比,ECNet的效率也提高了15-50 倍,而随机抽样是当前定向进化工作流程中广泛使用的一种策略。这些结果表明,ECNet是蛋白质工程检索高阶变体的有效方法,有可能提高实验室定向进化的效率。
4.从低阶变异数据归纳出高阶变异
构建和筛选高阶变异体需要大量的实验精力和时间。因此,与双突变体或高阶突变体相比,单突变体的适应性测量在现有的DMS研究中更为普遍。因此,在蛋白质工程中,人们非常希望根据低阶变异体的适配性数据训练的机器学习模型也能准确预测高阶变异体的适配性。这样,该模型就能充分利用筛选出的低阶变体的适配性数据,并优先选择可能在下一轮定向进化中表现出更好特性的高阶变体。研究人员评估了ECNet在仅使用低阶数据进行模型训练时预测高阶变体适配性的性能。研究人员从之前的DMS研究中收集了六个蛋白质的单突变体和双突变体的适应性测量结果。然后,研究人员仅使用单突变体数据训练预测模型,并测试其在双突变体上的性能。对于这六种蛋白质,ECNet的Spearman相关性从0.73到0.94不等,表现优于TAPE和EVmutation方法(图4a),这表明它可以从低阶变异数据中预测高阶变异。
实验中还观察到,训练数据中适配性景观多样性的增加提高了预测性能。例如,为了预测avGFP蛋白四突变体的适配性,使用单突变体、双突变体、三突变体或所有突变体的适配性数据分别训练了不同的模型。测试结果表明,使用高阶突变数据(从单到三)训练的模型预测效果越来越好,而结合全部突变数据则进一步提高了预测效果(图4b)。为了进一步评估ECNet的能力,研究人员使用了包含146个已知具有抑制剂抗性的TEM-1变异序列的正交数据。与TEM-1蛋白相比,这些数据中的序列包含2到10个(平均3.3个)氨基酸替换。在这些序列的基础上,列举了限制在146个变体中引入突变的位置的所有突变组合,从而生成了十倍以上的随机变体。然后,在TEM-1单一突变体数据的适配性数据上训练了模型,并用它来预测146个TEM-1变异体和随机生成的变体的适配性。
研究人员发现,ECNet将抗抑制剂变体与随机变体背景区分开来(图4c;平均预测适合度 0.79 vs. 0.48;单侧秩和检验 P1<10-5)。这种正交验证进一步证明了ECNet的通用性,即使是在单一突变体数据上训练的ECNet也能预测高阶突变体。
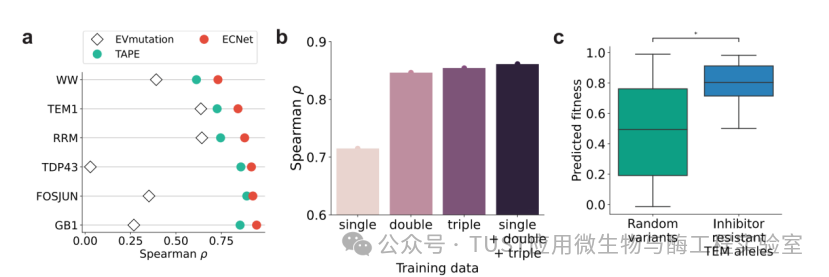
图4 使用在低阶变量上训练的模型准确预测高阶变量
5.利用 ECNet 工程化 TEM-1 β-内酰胺酶
为了在实验中验证其在蛋白质工程中的实用性,研究人员应用ECNet优先选择新的高阶TEM-1 β-内酰胺酶变体,这些变体与野生型相比可能具有更好的适应性。研究人员使用先前研究中报告的DMS数据训练了ECNet。这些数据集收集了几乎所有TEM-1单点突变体和12%可能的连续双突变变体的适应性测量结果。研究人员对文献中收集的TEM1的几个功能相关位点及其高阶重组进行了模拟诱变。然后,应用ECNet来预测由模拟诱变产生的所有变体的适配性。
在剔除结构不稳定的变体后,选出了37个在标准ECNet模型或ECNet组合版本(ECNet模型的多个重复预测的平均值)中排名靠前的变体。这37个名列前茅的变体是新的TEM-1变异体,与研究人员训练数据中的任何变体或研究人员从文献中收集的功能性TEM-1变异体都没有重叠。尽管训练数据只涵盖单突变体和连续双突变体,但这37个变体采样了不同的突变位点组合,并包含2到6个突变的高阶突变体。
研究人员创建了这37个变体和之前报道的9个对氨苄西林具有强抗性的TEM-1突变体作为阳性对照。将包含这37个变体和阳性对照的文库培养在含有不同浓度(300、1500和3000微克/毫升)氨苄青霉素的LB琼脂平板上,以测试它们对氨苄青霉素的抗性。此外,还进行了PacBio测序,以确定这些变体在选择前后的相对丰度,作为其适应性的代表(图5a)。每个突变体在特定氨苄青霉素浓度下的适应性是根据平板中含有相关浓度氨苄青霉素的突变体与野生型 TEM-1的相对丰度之比,以及平板中不含有氨苄青霉素的突变体与野生型TEM-1的相对丰度之比计算得出的。实验观察到,与野生型相比,ECNet 优先考虑的大多数变异体的适应性都有所提高(图5b)。
在不同浓度的氨苄青霉素(300、1500和3000 μg/mL)下都观察到了这种改善,并且在不同的重复中具有可重复性。值得注意的是,ECNet发现的变体可将野生型的适应性提高约8倍,大大高于训练数据中表现最好的变体(图5b中的阳性对照)。研究人员还发现,ECNet的集合模型实现了稳健的预测,在浓度为300、1500和3000 μg/mL的情况下,平均命中率(预测变体的适合度高于野生型的比例)分别为 0.52、0.91 和 0.94(图5c)。
尽管ECNet是在单突变体和连续双突变体的数据基础上进行训练的,但它优先选择了对氨苄青霉素耐药性更强的新型高阶TEM-1突变体。验证结果表明,进化背景使ECNet能够发现训练数据中未观察到的高阶突变体。这些结果还证明了ECNet可以集成到现有的蛋白质工程工作流中,指导发现性能提升的变体。
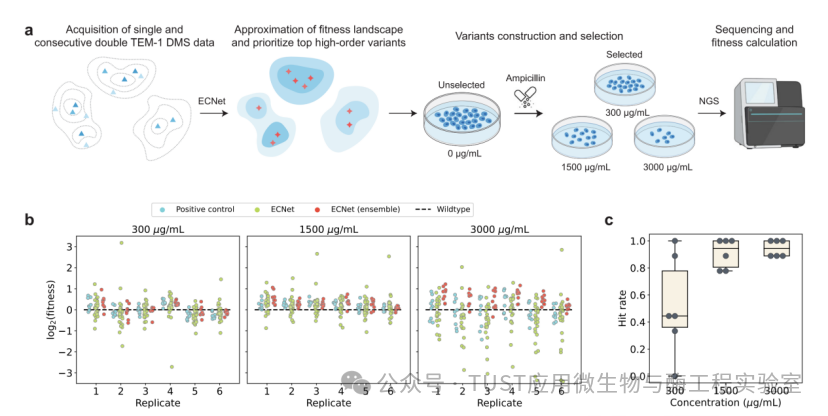
图5 ECNet使TEM-1的快速工程化成为可能









