1 TRIAGE R包助力基因调控研究,揭示细胞行为的奥秘
❝文章:TRIAGE: an R package for regulatory gene analysis
链接:https://academic.oup.com/bib/article/26/1/bbaf004/7952010
代码:https://github.com/palpant-comp/TRIAGE_R_Package
基因调控是理解细胞发育、功能以及应对疾病的核心,但传统的RNA测序(RNA-seq)方法在揭示调控网络时存在局限。为此,昆士兰大学的研究团队开发了TRIAGE R包,为基因调控研究提供了全新解决方案。
TRIAGE全称为“基于表观遗传学的转录调控推断分析”(Transcriptional Regulatory Inference Analysis Guided by Epigenetics)。它通过利用H3K27me3数据(与特定DNA调控区域相关的表观遗传标记),从RNA-seq数据中挖掘调控元件,揭示基因调控网络和细胞行为的奥秘。TRIAGE适用于批量和单细胞RNA-seq数据,能够无缝集成到现有分析流程中。
TRIAGE的设计初衷是与现有的RNA-seq分析流程无缝集成,无论是批量RNA-seq数据还是单细胞RNA-seq数据都能适用。它的核心功能包括:
- 识别细胞类型特异性区域:通过H3K27me3数据,TRIAGE能够增强对特定细胞类型独特调控区域的分析。
- 揭示调控网络:TRIAGE可以揭示调控基因之间的相互作用,帮助科学家了解驱动细胞身份和功能的分子通路。
- 定制化分析流程:TRIAGE支持用户根据自己的研究目标调整分析流程,适应多种数据集和研究需求。
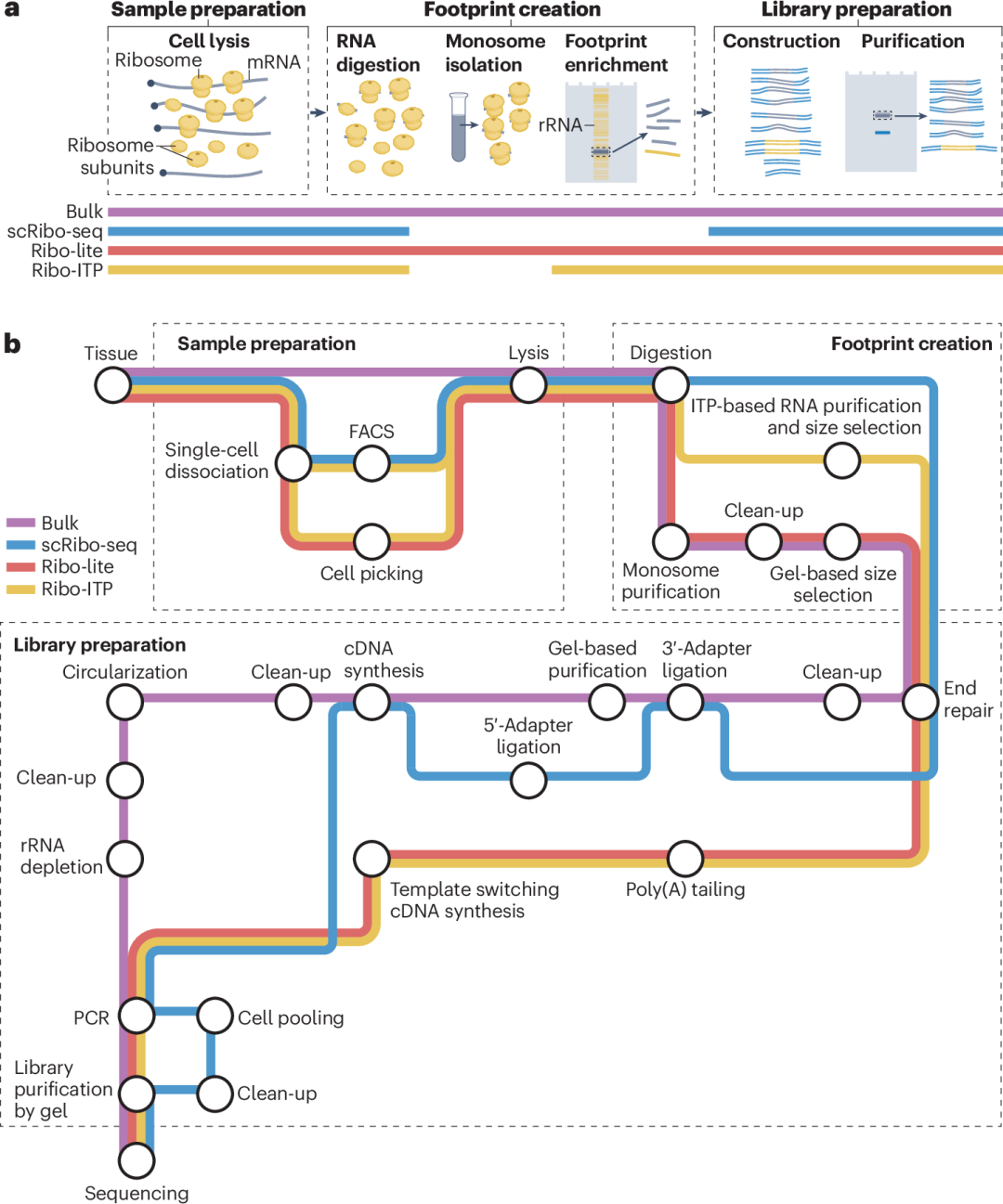
TRIAGE R包提供了一系列强大的功能,帮助研究人员更高效地分析和可视化数据。例如:
- plotJaccard函数:以热图的形式可视化不同基因组群体之间的Jaccard相似性指数。
-
compareGO函数:比较不同基因集合的GO(基因本体)富集情况,并通过点图展示富集模式。
- topGenes函数:识别每个群体中TRIAGE加权值最高的基因。
- TRIAGEcluster模块:通过识别更具体的“TRIAGE峰”优化单细胞RNA-seq数据的细胞聚类。
- TRIAGEparser模块:解析基因列表或表格,识别基因簇及其基因本体。
研究团队在三个不同的转录组数据集上测试了TRIAGE R包,结果表明,它能够轻松集成到标准工作流中,并揭示驱动多种生物过程(从发育到疾病)的调控机制。这些应用展示了TRIAGE在研究细胞行为和疾病机制中的巨大潜力。
2 singleDeep:一款专为单细胞RNA测序数据设计的深度学习分析工具
❝文章:Explainable deep neural networks for predicting sample phenotypes from single-cell transcriptomics
链接:https://academic.oup.com/bib/article/26/1/bbae673/7954570
代码:https://github.com/GENyO-BioInformatics/singleDeep
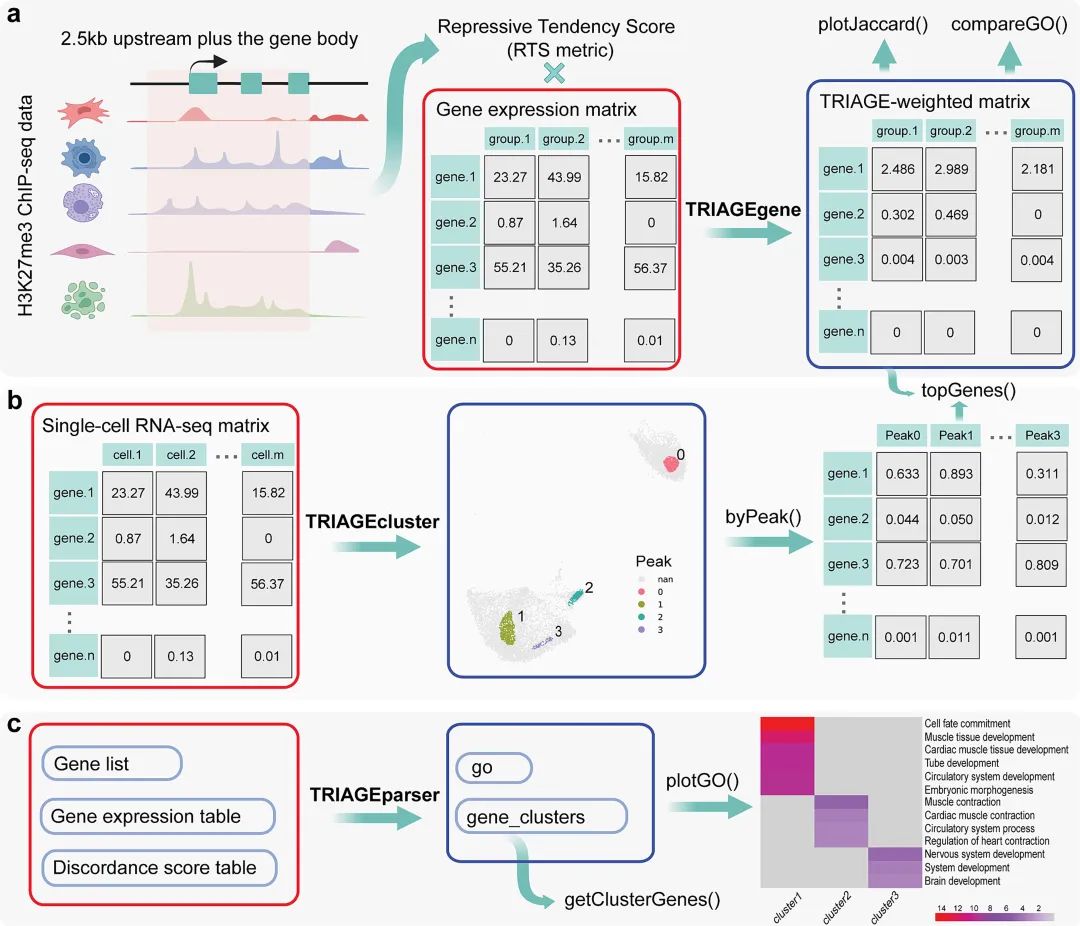
传统的基因表达分类方法通常依赖于样本的基因活性模式进行分组。这种方法在处理普通数据时表现良好,但面对单细胞数据的高度复杂性和细胞间的巨大变异性时显得捉襟见肘。为了应对这些难题,GENYO(基因组与肿瘤学研究中心)的研究团队开发了singleDeep,一款专为单细胞RNA测序数据设计的深度学习分析工具。
singleDeep是一个端到端的分析流程,能够从单细胞RNA测序数据中提取基因表达矩阵,并结合每个细胞群的注释信息进行深度学习建模。它可以为每个细胞群体训练专属的神经网络,并通过内部交叉验证(nested CV)生成训练报告、基因贡献分析和性能指标。更重要的是,singleDeep还能利用预训练模型对外部数据进行预测,从而实现对新样本表型的精准分类。
singleDeep已经在多种疾病研究中展现出强大的应用潜力。例如,在系统性红斑狼疮的研究中,singleDeep成功识别出与自身免疫相关的特定干扰素标志基因,这些基因在不同免疫细胞类型中表现出一致性。在阿尔茨海默病研究中,它揭示了APOE等基因在星形胶质细胞等脑细胞群体中的关键作用,为理解痴呆的分子机制提供了新视角。此外,singleDeep还被用于COVID-19研究,表现出卓越的诊断能力,其预测结果显著优于现有方法。
3 Canopy2工具助力肿瘤异质性研究新突破
❝文章:Canopy2: Tumor Phylogeny Inference by Bulk DNA and Single-Cell RNA Sequencing
链接:https://link.springer.com/article/10.1007/s12561-024-09466-1
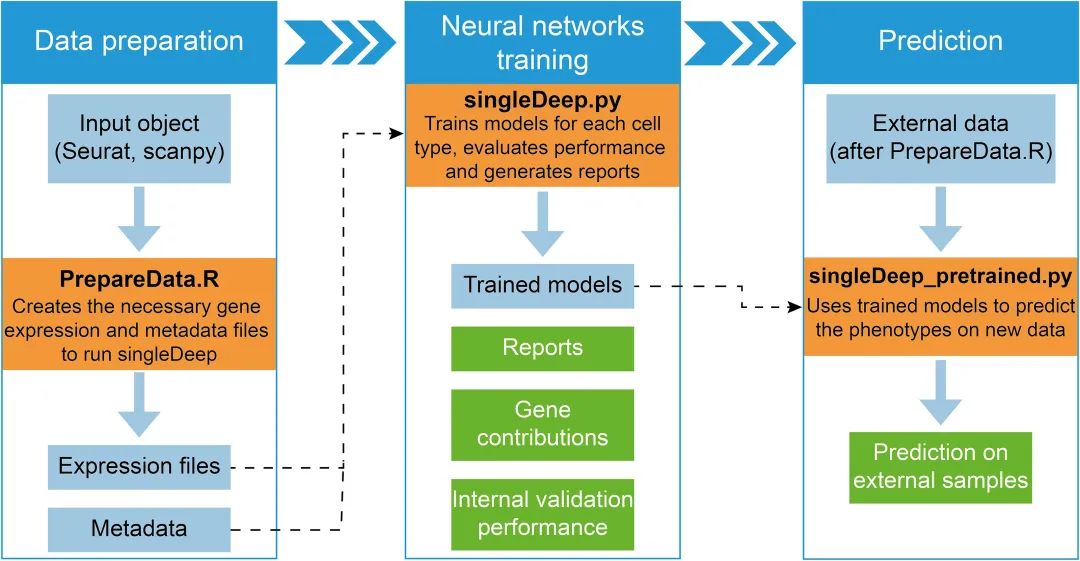
癌症并不是由一种单一类型的细胞组成,而是由各种遗传和功能上截然不同的细胞混合而成。这种被称为“肿瘤异质性”的现象是癌症治疗失败的重要原因之一,因为肿瘤细胞在治疗过程中可能发生进化,产生对药物的抗性。要理解这种复杂性,科学家们需要构建肿瘤细胞的“家谱”——也就是肿瘤谱系树,来揭示不同肿瘤细胞的进化过程。然而,绘制这样一棵谱系树绝非易事,特别是在数据稀疏或存在测序噪声的情况下。为了解决这一难题,北卡罗来纳大学教堂山分校的研究人员开发了一款名为Canopy2的工具,为肿瘤研究提供了全新的突破口。
Canopy2 是一个基于贝叶斯统计框架的工具,专门设计用于分析肿瘤的遗传数据。它结合了大规模 DNA 测序和单细胞 RNA 测序两种技术,能够精确重建肿瘤细胞的进化历史。肿瘤谱系树的构建依赖于单核苷酸变异(SNV)的数据,这些变异是肿瘤细胞进化过程中积累的遗传标志。Canopy2 能够将这些变异整合起来,绘制出肿瘤细胞的“进化地图”。例如,在仅使用大规模 DNA 数据时,虽然可以推断突变的时间顺序,但难以准确还原肿瘤谱系的分支结构。而单细胞 RNA 数据虽然能够提供更清晰的分支结构,却因为测序噪声和缺失值而容易导致时间顺序的错误。Canopy2 的强大之处在于能够整合这两种数据,弥补各自的不足,从而构建更全面、更准确的肿瘤谱系树。
研究人员使用 Canopy2 对一些具有挑战性的肿瘤样本进行了测试,包括乳腺癌和胶质母细胞瘤。这些肿瘤通常具有高度的异质性,并且在晚期阶段会出现多种不同类型的突变细胞。实验结果表明,Canopy2 能够在这些复杂条件下准确重建肿瘤的克隆谱系树(即肿瘤细胞的“家谱”),并优于现有的其他方法。例如,在乳腺癌的分析中,Canopy2 成功区分了不同亚群的肿瘤细胞,并揭示了某些关键突变的时间顺序和分支结构。这些信息对于理解肿瘤的进化过程至关重要,也为开发针对性的治疗策略提供了科学依据。
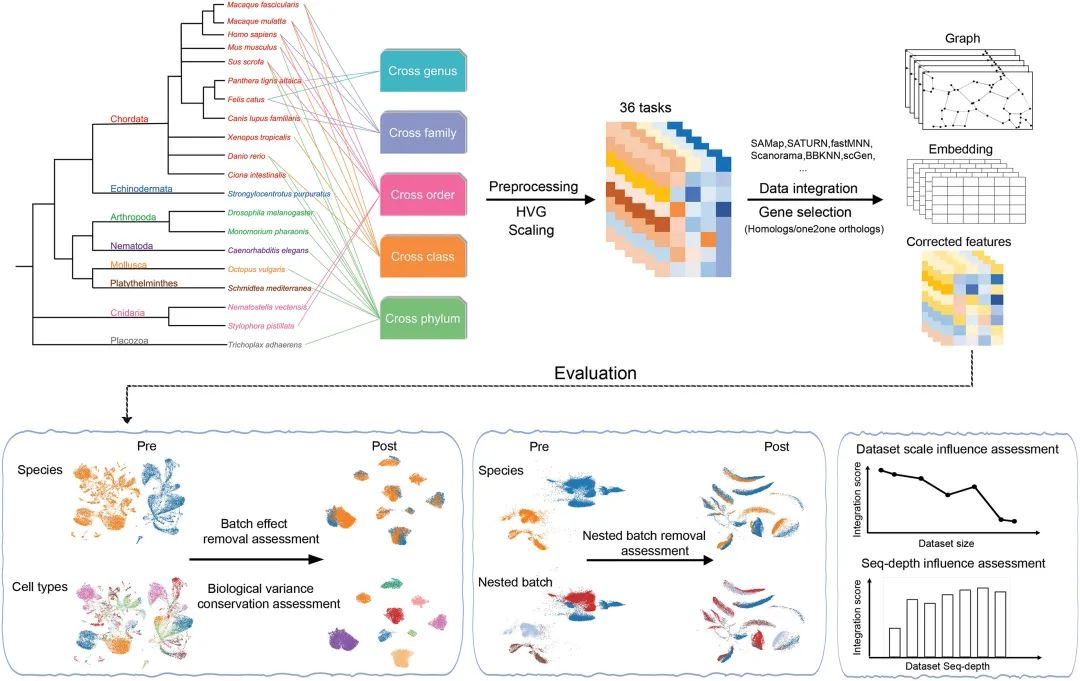
4 跨物种单细胞RNA测序数据整合方法迎来突破
❝文章:Benchmarking cross-species single-cell RNA-seq data integration methods: towards a cell type tree of life
链接:https://academic.oup.com/nar/article/53/1/gkae1316/7945393
跨物种单细胞RNA测序研究的核心在于比较不同物种的细胞类型和功能,但这并非易事。每个物种都有独特的基因序列,这使得直接比较它们的基因表达变得复杂;不同实验室的样本制备和测序方法差异则会引入“批次效应”,即数据中的人为差异;即便是同一物种的个体之间,也可能因年龄、环境或健康状况的不同而表现出显著的生物学差异。这些挑战往往掩盖了跨物种研究中真正有意义的生物学模式。
为了克服这些问题,KAUST研究团队从20种动物中收集了470万个细胞的数据,涵盖了8个主要动物门类(phyla)。他们设计了36个跨物种数据整合任务,测试了包括SATURN、SAMap和scGen在内的9种主流整合方法,并使用13个指标评估它们的表现,主要关注去除批次效应和保留生物学差异。此外,他们还特别考察了在数据不平衡(某些物种数据较少)和测序深度低的情况下,这些方法的表现。
研究结果显示,不同的数据整合方法在去除批次效应和保留生物学差异方面各有优劣,适用于不同的研究目标和场景。基因序列驱动的方法利用基因序列的相似性对齐数据,擅长保留生物学差异,非常适合探讨进化关系的研究;生成模型(Generative Models)通过复杂的数学模型去除批次效应,使数据更为一致,适合需要清晰数据集的下游分析。其中,SATURN、SAMap和scGen三种工具表现尤为突出:SATURN在从近缘属到远缘门的广泛分类等级中表现优异,是整合多样化数据集的通用选择;SAMap在处理非常远缘物种(如跨科或跨门的比较)时表现最佳,适合大规模的进化研究;scGen则在研究较近缘物种(如同一纲内的比较)时效果显著,是小范围整合的理想工具。
这项研究不仅为科学家们提供了关于整合方法的清晰指南,还强调了在选择工具时需要根据研究目标和数据特点做出权衡。例如,如果研究目标是探索物种之间的广泛进化关系,推荐使用SATURN或SAMap;如果研究集中于近缘物种(如同一科内的物种),scGen可能是更好的选择;如果需要同时平衡批次效应去除和生物学差异保留,则可以根据具体数据特点选择最合适的方法。
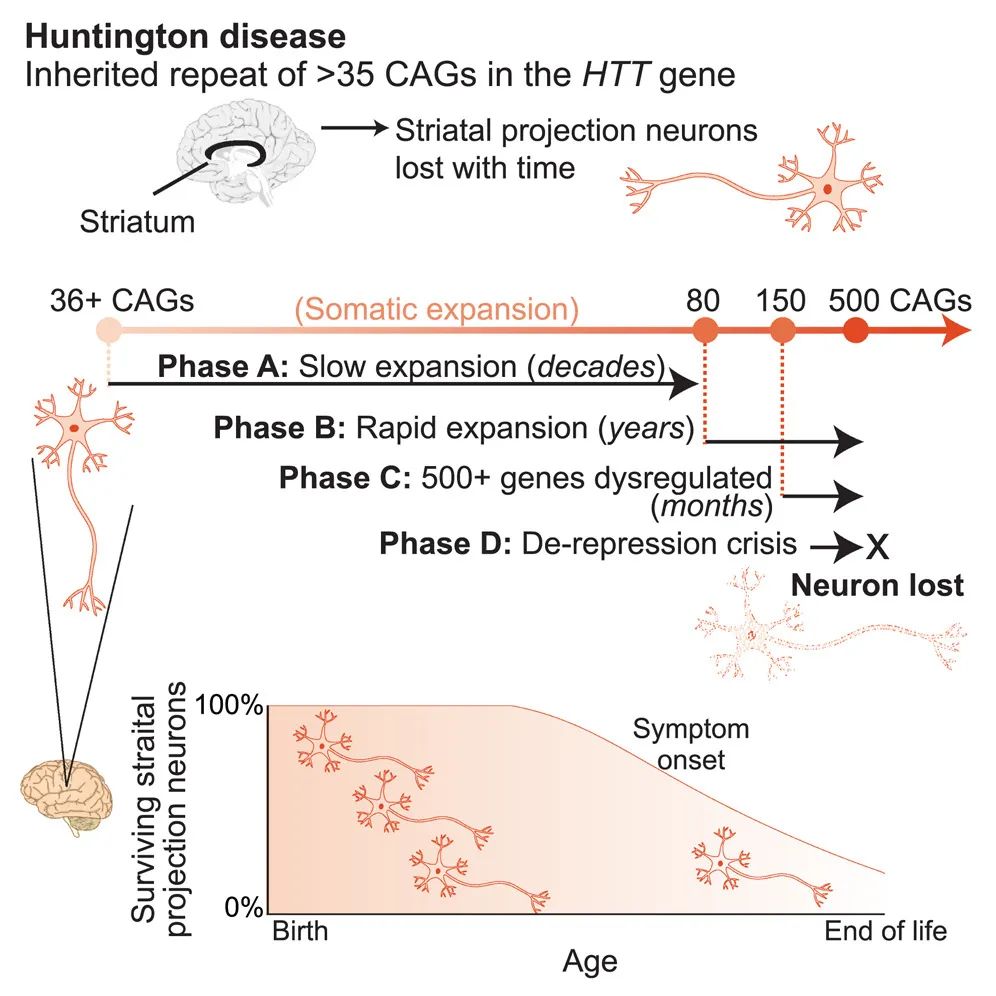
5 亨廷顿病新突破:科学家揭示致病关键机制,找到延缓疾病的潜在方法
❝文章:Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s diseaseLong somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease
链接:https://www.cell.com/cell/fulltext/S0092-8674(24)01379-5
亨廷顿病,这种令人闻之色变的遗传性神经退行性疾病,长期以来困扰着科学界。尽管早在1993年科学家就发现了导致该病的基因突变——Huntingtin(HTT)基因中的CAG重复序列扩增,但这一突变究竟如何杀死脑细胞,为什么患者在中年才发病,始终是未解之谜。然而,近日,麻省理工学院-哈佛大学布罗德研究所、哈佛医学院和麦克莱恩医院的科学家们在《Cell》杂志上发表了一项重磅研究,首次揭示了亨廷顿病的核心致病机制,并提出了全新的治疗思路。研究表明,亨廷顿病的基因突变并不会直接伤害细胞。相反,这种突变在患者生命的前几十年几乎是“无害”的,但随着时间推移,突变的DNA重复序列逐渐扩增,直到达到一个关键阈值(约150个CAG重复序列),才会引发细胞的基因表达紊乱并导致细胞死亡。更令人震惊的是,这种扩增现象主要发生在大脑中某些特定的神经元——纹状体投射神经元中,而其他细胞几乎不受影响。这一发现颠覆了科学家对亨廷顿病的传统认知。
研究团队通过分析53名亨廷顿病患者和50名健康个体的大脑组织,结合先进的单细胞RNA测序技术(Drop-seq),测量了超过50万个单细胞的基因表达和DNA重复序列长度。他们发现,大多数细胞的CAG重复序列长度与遗传的突变一致,但纹状体投射神经元中的CAG重复序列显著扩增,甚至达到800个重复。这种扩增过程在患者生命的前20年非常缓慢,每年仅增加1次左右,但当重复序列达到约80时,扩增速度显著加快,在几年内迅速达到150,随后细胞在几个月内死亡。正是这些细胞的逐渐消失,导致了患者运动、认知和吞咽功能的逐步衰退。
这一发现还解释了为何现有针对HTT蛋白的治疗效果有限。由于在任何时间点,只有极少数细胞拥有有毒版本的HTT蛋白,这些疗法无法显著改善患者整体的病情。研究团队提出,与其仅仅抑制HTT蛋白的表达,不如尝试阻止或减缓CAG重复序列的扩增,这可能会延缓疾病的进展甚至预防疾病的发生。
此外,研究还揭示了DNA修复蛋白(如MSH3)在这一过程中扮演的重要角色。MSH3本应帮助细胞修复DNA,但在面对额外的CAG重复序列时,它可能被“误导”而促进了扩增过程。通过调节这些蛋白的活动,或许可以减缓扩增速度,为患者争取更多健康时间。
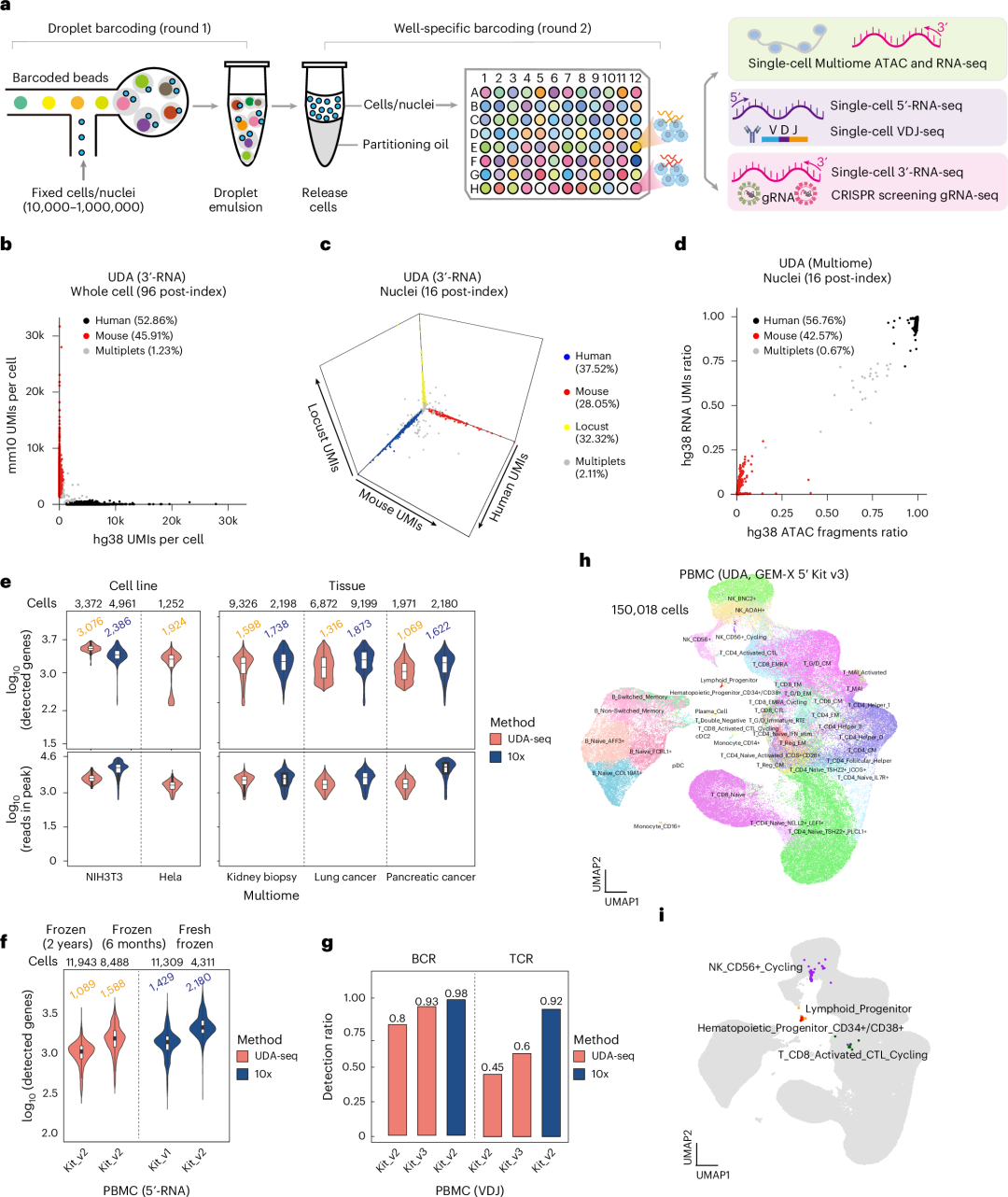
6 单细胞测序新突破:UDA-seq技术实现高效、低成本、多模态数据获取
❝文章:UDA-seq: universal droplet microfluidics-based combinatorial indexing for massive-scale multimodal single-cell sequencing
链接:https://www.nature.com/articles/s41592-024-02586-y
现有的单细胞RNA测序技术虽然提供了细胞层面的分子信息,但在区分功能相似却分子特性不同的细胞类型时存在局限性。而目前的多模态测序方法虽然能整合更多维度的信息,但其高昂的成本和较低的通量限制了广泛应用。近日,一项发表在《Nature Methods》上的研究提出了一种名为UDA-seq(Universal Droplet-based Combinatorial Indexing for Single-cell Sequencing)的全新技术,通过通用微流控组合索引方法,大幅提升了单细胞多模态测序的规模和性价比。这一突破性进展为单细胞研究开辟了新的可能性。
UDA-seq技术通过两轮组合索引,结合微流控技术和后续PCR扩增步骤,能够生成多达9.6至38百万个条形码组合。该方法不仅适用于RNA测序,还兼容VDJ测序、染色质开放性分析以及CRISPR筛选等多种测序需求。
研究团队对这一技术进行了验证,在一次实验中成功分析了35份冷冻临床样本,获得了超过10万个高质量单细胞数据集。更为重要的是,该方法在检测稀有细胞群体方面表现出色,例如发现了与衰老相关的γδT细胞(占PBMC的0.36%)以及与蛋白尿相关的足细胞(占细胞总数的不到0.6%)。此外,UDA-seq在单细胞RNA检测率上与现有标准方法相当(每个细胞平均检测到1707个基因),但每个样本的成本却降低了8到20倍。
这一技术的核心优势在于其灵活性和可扩展性。通过固定细胞或细胞核、微滴条形码标记、细胞释放和多孔板分装等步骤,UDA-seq为现有的单细胞多模态方法提供了一种通用、低成本的解决方案。研究团队表示,这一技术不仅适用于基础研究,还将推动单细胞技术在临床应用中的普及,为疾病机制研究、药物开发和个性化医疗提供更强大的工具。
7 单细胞翻译测序新突破:解锁基因表达的“最后一公里”
❝文章:Sequencing technologies to measure translation in single cells
链接:https://www.nature.com/articles/s41580-024-00822-z
翻译是基因表达的关键步骤之一,也是细胞中最耗能的过程之一。尽管科学家们已经开发了多种技术来研究翻译调控,但在多细胞组织、稀有细胞类型和动态生物过程中,翻译调控的研究仍然面临技术瓶颈。近日,《Nature Reviews Molecular Cell Biology》发表了一篇综述文章,聚焦单细胞翻译测序技术的最新进展。这些技术的出现,不仅让我们能够在单细胞水平上研究翻译过程,还为揭示翻译调控在发育、疾病及癌症中的作用提供了全新视角。
在传统的基因表达研究中,科学家们主要依赖测量mRNA水平的单细胞转录组测序(scRNA-seq)来解析细胞类型和状态。然而,mRNA的丰度并不能直接反映蛋白质的实际合成水平,因为翻译调控在基因表达中起着重要作用。为了解决这一问题,研究者们开发了多种单细胞翻译测序技术,包括核糖体测序、核糖体亲和纯化以及空间翻译测序等。这些技术的共同目标是从单细胞中捕捉翻译活动的全貌。
核糖体测序(Ribosome Profiling)是目前最经典的翻译测量方法。其原理是通过检测被核糖体保护的mRNA片段(即“核糖体足迹”),精确定位核糖体在转录本上的位置和数量。这些数据可以揭示翻译起始、延伸和终止的动态过程。然而,传统核糖体测序需要大量样本,难以应用于单细胞研究。为此,科学家们进行了多项技术优化,例如单细胞核糖体测序(scRibo-seq)将所有处理步骤整合到单管反应中,大幅提高了灵敏度和通量。这一技术已被用于研究细胞周期中的翻译动态以及稀有细胞类型的翻译特性。
另一种方法是核糖体亲和纯化(Ribosome Affinity Purification),通过拉取与核糖体结合的mRNA来评估翻译效率。例如,T&T-seq技术通过分离单细胞裂解液的一部分进行转录组测序,另一部分用于核糖体结合mRNA的纯化。这种方法能够同时测量单细胞的转录组和翻译组,为研究翻译调控与mRNA丰度之间的关系提供了宝贵的工具。然而,这种方法需要分离样本,限制了其在小细胞样本中的应用。
最令人兴奋的进展之一是空间翻译测序技术(如RIBOmap)。这一技术结合了空间转录组学的优势,可以在组织切片中测量核糖体结合的mRNA的空间分布。通过这种方法,科学家们发现了不同细胞类型或亚细胞区域中翻译活动的显著差异。例如,在小鼠脑组织中,神经元细胞的翻译活动主要集中在细胞体,而非神经元细胞则表现出更强的翻译调控能力。这一发现为研究局部翻译如何影响蛋白质功能提供了全新视角。
尽管这些技术为单细胞翻译研究带来了革命性进展,但它们仍处于发展的早期阶段。现有方法在通量、灵敏度和数据处理复杂性方面仍有改进空间。例如,核糖体测序的样本制备步骤较为繁琐,难以与现有的商业化单细胞转录组测序平台兼容。此外,由于翻译调控过程的动态性,如何在单细胞水平上同时测量转录组和翻译组仍是一个技术难题。