近年来,机器学习原子间势能(MLIPs)的研究取得了显著进展,为大规模分子动力学模拟提供了一种高效且准确的替代方案。与传统的量子力学方法相比,MLIPs通过拟合量子力学势能面,可在接近量子精度的前提下实现计算效率的大幅提升。然而,现有MLIPs多局限于特定体系或少数元素,通用性不足。
为解决这一问题,卡内基梅隆大学研究团队开发了第二代原子-分子神经网络势能模型(AIMNet2),该模型通过整合机器学习与物理基础的长程相互作用项,能够处理包含14种非金属元素的中性及带电分子体系,实现了从基础有机分子到具有复杂元素有机化合物的通用建模。近日,该项研究工作发表在英国皇家化学学会出版的Chemical Science期刊【1】。
AIMNet2的架构(图-1)结合了机器学习参数化的短程作用与物理驱动的长程相互作用项。模型输入为原子坐标、原子序数和体系净电荷,模型采用消息传递方法,通过卷积操作结合原子和几何描述符,计算原子特征向量。AIMNet2的总能量计算由三个部分组成,包括局部组态相互作用能量(local configurational interaction energy)、显式色散校正项(explicit dispersion correction)及原子中心部分点电荷间的静电作用。其中,局部组态相互作用能量通过分子中原子层(Atoms-in-molecules, AIM)计算。
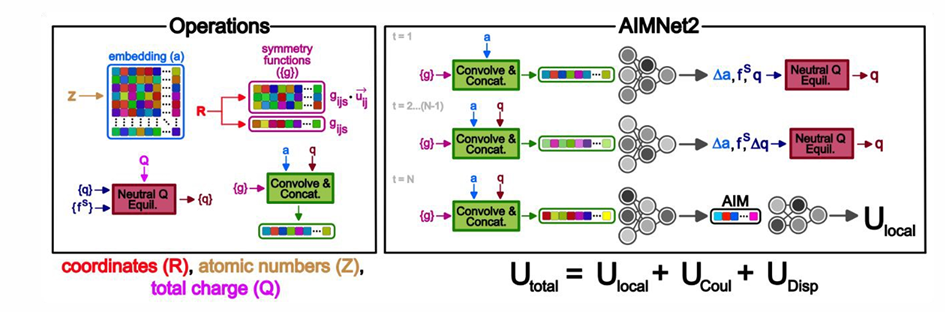
图-1: AIMNet2架构图
AIMNet2的AIM层负责从分子体系的局部化学环境中提取原子级特征表示,通过多轮消息传递生成每个原子的特征向量(AIM向量),该向量编码了原子周围的化学环境信息,用于计算局部能量项。该层以原子坐标、元素类型及体系总电荷为输入,利用径向对称基函数和原子嵌入矩阵编码原子间距离与电荷分布,结合神经电荷平衡(NQE)动态调整分子电荷分布,以适应带电体系。
AIM层专注于短程相互作用,与显式长程静电和色散项互补,兼顾局部键合与非键相互作用的精确建模。
在数据方面,训练通用型MLIPs的核心挑战在于数据集的规模与质量,主数据集的分子结构来自PubChem、ChEMBL等数据库,通过构象采样、元动力学等方式,获得约1.2亿个分子构象,每个样本通过低精度DFT方法标注能量和原子力。研究团队通过数据蒸馏技术,从主数据集中筛选出2000万个最具信息量的样本,用于模型训练。具体过程如图-2所示,首先随机选取10万个样本训练初始AIMNet2模型;再遍历主数据集,选择能量或原子力预测误差超过当前模型平均训练误差的3倍的“高信息量样本”加入训练集,逐步提升模型覆盖能力。当筛选后的训练集规模达到约2000万样本时,模型可准确预测主数据集全部样本。模型通过误差阈值动态筛选“难样本”,优先学习模型未掌握的特征,压缩冗余数据,从而提高训练效率。
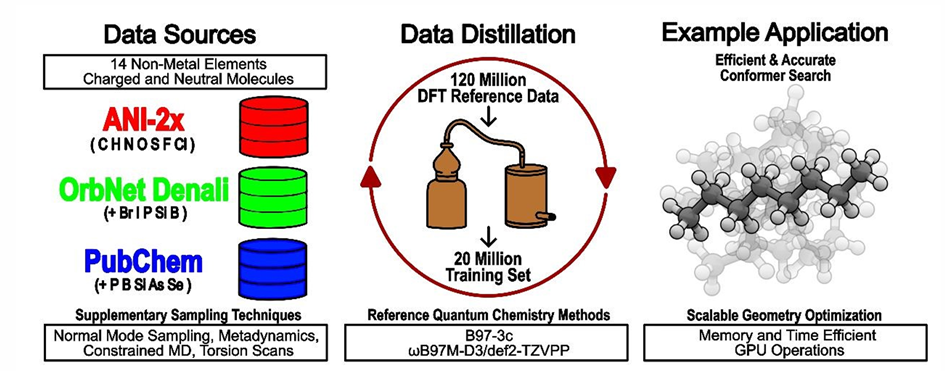
图-2: 数据蒸馏过程
完成数据蒸馏后,对筛选后的核心样本采用高精度ωB97M-D3/def2-TZVPP方法重新计算能量与原子力,确保关键数据的可靠性。基于此从头训练4个独立模型,通过集成学习消除单模型偏差,提升预测稳定性与泛化能力。
研究人员通过多项基准测试验证了AIMNet2的可靠性与通用性。
在非常规的化学键几何优化方面,研究人员从剑桥结构数据库中选取113个含稀有化学键的分子(如六配位氯离子、硒掺杂硼簇),AIMNet2优化后的几何结构与实验晶体结构的平均RMSD为0.38 Å,优于半经验方法GFN2-xTB。
在构象搜索任务中,研究人员选择676个含1-3个可旋转键的分子,评估在未知实验结构的情况下,模型能否从生成的构象池中筛选出结构正确且能量稳定的候选构象。如图-3所示,AIMNet2识别实验构象的成功率达77%,与直接使用B97-3c泛函的结果(75%)相当,显著高于半经验方法GFN2-xTB(45.2%)。
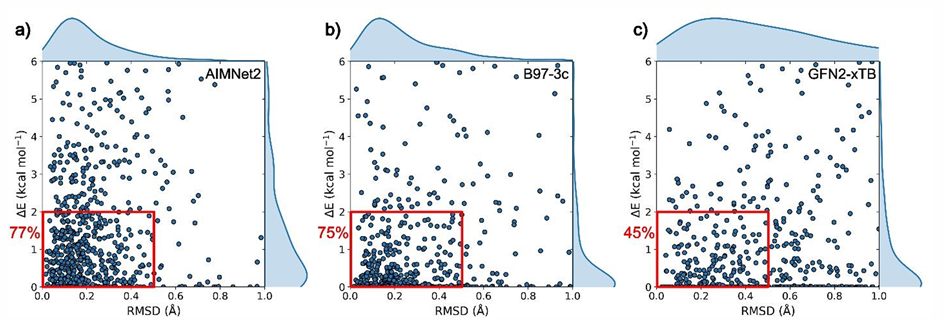
图-3: 不同方法构象识别成功率
如图-4b所示,AIMNet2在氢键(HB)、Sigma-hole相互作用(SH)及离子氢键(IHB)中误差显著低于GFN2-xTB,接近DFT精度;色散(D442)因长程特性误差略高,但仍优于半经验方法。AIMNet2在复杂非共价作用中表现良好,但在色散体系仍需结合物理修正或扩展训练数据以进一步提升精度。
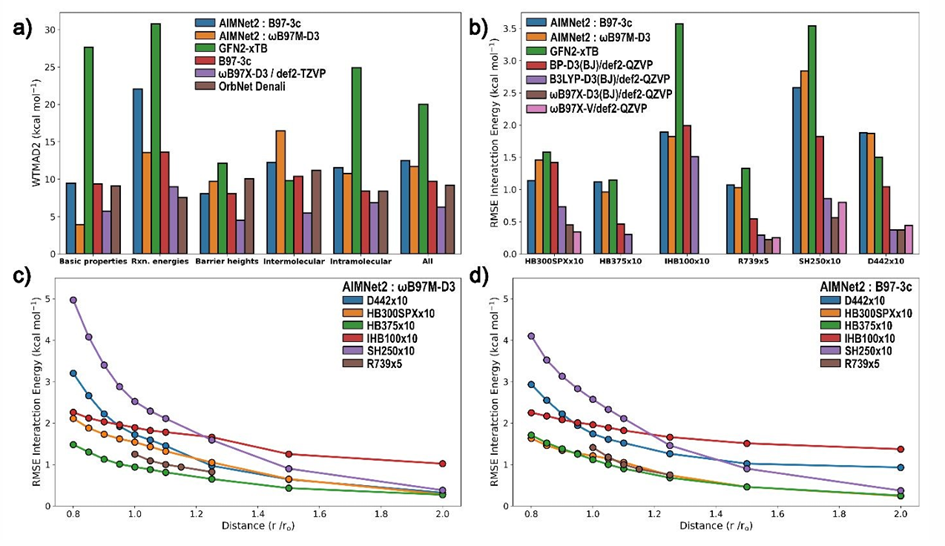
图-4 不同模型在非共价作用预测的性能
而在大分子与凝聚态模拟测试中,AIMNet2在GPU加速下优化含80个原子的分子构象比GFN-FF快5倍,且模型可扩展至10⁵原子规模的体系。分子动力学模拟显示,AIMNet2能稳定模拟1000个CO
₂分子的凝聚相2.5 ns。
小结:
AIMNet2通过融合机器学习参数化的短程作用项与物理理论长程作用项,在包含2×10⁷个DFT数据的训练下,实现了通用型MLIPs的突破性进展。其架构创新体现为:(1)显式引入长程作用,突破消息传递的局部性限制;(2)兼容中性与带电态;(3)支持14种元素。实验结果表明,该模型在相互作用能预测、构象搜索、大分子优化等任务中,性能超越GFN2-xTB并与基准DFT相当,可作为多数场景下DFT的高效替代方案。但是由于训练集主要针对小分子和简单非共价复合物,因此无法保证其在蛋白质等生物大分子领域的可靠性。
参考文献
[1] Olexandr Isayev, Dylan Anstine, and Roman Zubaiuk. AIMNet2: A Neural Network Potential to Meet your Neutral. Chem. Sci. 2025. https://doi.org/10.1039/D4SC08572H







