今天为大家带来的是发表在NATURE COMMUNICATIONS上的一篇文章,Improved protein structure refinement guided by deep learning based accuracy estimation,它提出了一个深度学习框架—DeepAccNet,通过估计蛋白质模型中的每个残基精度和每个残基-残基距离符号误差来指导Rosetta蛋白质结构预测模型的优化。
摘要
DeepAccNet中使用三维卷积来评估局部原子环境,之后使用二维卷积提供原子的全局背景。DeepAccNet在 Rosetta细化协议的多个阶段纳入准确性预测,大大提高了所得蛋白质结构模型的准确性。该网络可以广泛用于评估预测结构模型、实验确定的结构精度以及识别可能存在误差的特定区域。
1.介绍
基于氨基酸共同进化数据的距离预测已经显著促进蛋白质结构预测,但是在大多数情况下,预测结构依然与实际结构有很大差异。蛋白质结构改进的挑战在于提高此类初始模型的准确性。
采样是改进模型的主要困难。即便是在初始模型的附近,需要搜索的可能结构的空间也是巨大的。如果能够做到精确识别输入蛋白质模型的哪些部分最有可能发生错误以及这些区域该如何改变,那么就有可能很大程度上改善结构空间的搜索过程,从而改进整个优化过程。模型精度评估方法(EMA)有很多,包括像ProQ3D、Ornate这种基于深度学习的方法,也有非深度学习的方法。这些方法着重于预测每个残基的准确性,很少有基于深度学习的精度预测来指导改进的研究。
文章提出了一个基于深度学习的框架(DeepAccNet),该框架可以用来估计蛋白质模型中每个残基-残基距离中的符号错误以及局部残基接触误差,并使用此估计值来指导基于Rosetta的蛋白质结构模型优化。
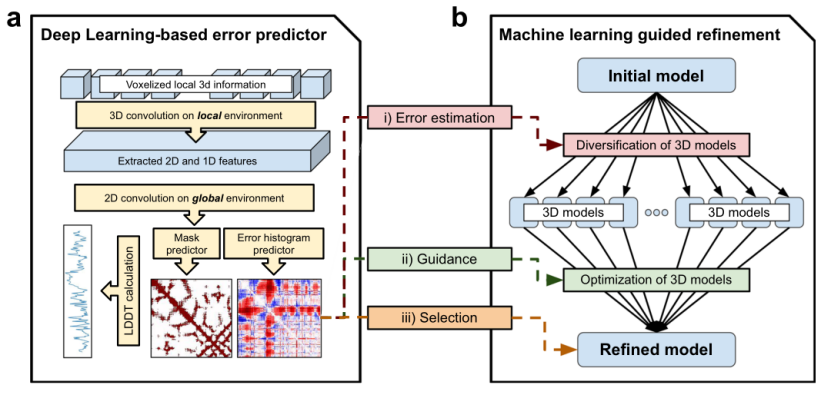
图1.DeepAccNet框架概览
2.方法
2.1 数据准备
通常蛋白质模型结构的训练集和测试集称为诱饵,相关的诱饵结构应该满足:拥有在序列空间中不太远或太近的模板、与其他的蛋白链没有很紧凑的接触、包含最小波动区域。文章选择了最大序列冗余为40%、最小分辨率2.5Å的20399个晶体结构,之后通过限制序列的大小在50-300个残基并且蛋白质要么是单体要么与其他链的相互作用很小,晶体结构个数减少至8718个。文章按照序列同一性不超过40%、序列覆盖率不低于50%等要求选取了50个模板进行模型生成。
文章通过同源建模、原生结构扰动和深度学习引导折叠三种方式进行诱饵结构生成。
同源建模:对于每个蛋白质,重复RosettaCM500次,每次从序列列表中随机选取一个模板。
原生结构扰动:通过扰动产生高精度的诱饵,利用RosettaCM通过两种方式产生30个模型,一是将原生结构的一部分模型与高精度模板结合,二是在原生结构的随机位置插入片段。
深度学习引导折叠:该方法利用trRosetta,对每个蛋白质进行五次不同深度的子采样多序列比对,标准的trRosetta建模对每个子采样的MSA运行45次。
对于每一个蛋白质,最终生成的诱饵结构共150个,三种方式分别生成90、30、30个诱饵。
2.2 模型结构和输入特征
文章中的框架在多个维度上进行卷积运算,不同类别的特征在网络的不同入口点进入。网络的第一组输入特征是每个残基中原子的体素化笛卡尔坐标。在由骨干N、Ca和C原子定义的相应局部坐标框架中,对每个残基单独进行体素化。第二组输入是每个残基的一维特征和每个残基对的二维特征。
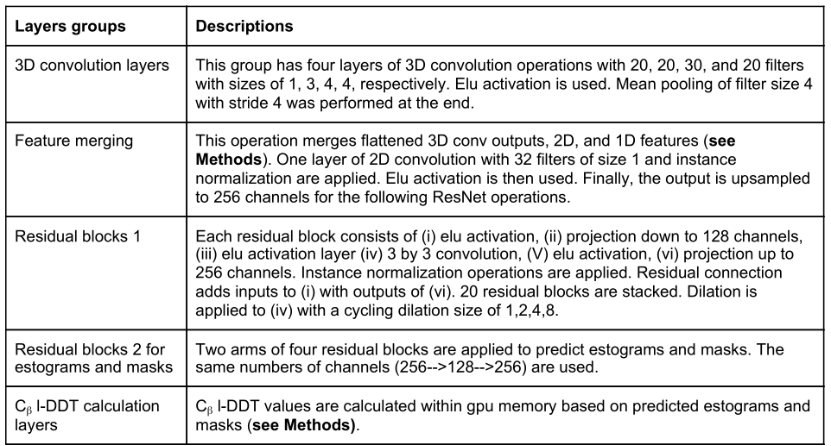
3D convolution layers:体素化原子坐标经过一系列的三维卷积层,其参数在各残基间共享,这些卷积运算生成描述蛋白质中每N个残基的局部三维环境特征。由该卷积层产生的输出经过扁平化处理成为每个残基的一维向量并与其他一维特征进行连接。
Feature merging:匹配特征维数并进行二维卷积运算。通过特征矩阵运算,每对残基既有该对残基的二维输入特征,又有两个单独残基的一维特征这种数据表示可以对骨干链和成对的相互作用进行卷积。
Residual blocks:连接后的特征矩阵进入一个残块数量为20的残差网络,之后网络分支成两部分,分别用来预测所有残基对的Cβ距离误差分布(estograms)和阈值为15Å的Cβ接触图(masks)。根据estograms和masks的概率分布预测,计算每个残基Cβ的局部距离差异测试(l-DDT)分数。
2.3 指导改进
本研究整体框架包括两个阶段:首先是初始模型多样化阶段,然后是迭代模型强化阶段,在该优化过程中,通过进化算法来维护结构池。
多样化阶段:继单一起始模型的精度估计之后,使用RosettaCM尝试2000个独立的Rosetta建模。迭代强化阶段:对一系列精度估计、新结构生成和池选择步骤进行迭代。在每次迭代中,从当前池中选择10个模型结构,然后对每
10个结构进行单独的精度预测,以指导从每个结构开始生成12个新的模型结构。从50个以前的和120个新生成的池成员中选择大小为50的新池,选择标准为估计的全局Cβ l-DDT最高和池内模型多样性。该过程迭代50次,在每5次迭代中,根据网络预测的残基Cβ l-DDT值,将模型结构与池中的另一个成员进行一次重组。2.4 最终的模型选择
在50个最终池成员中选出一个具有最高预测全局Cβ l-DDT分数的模型,然后从整个迭代细化轨迹中收集与该结构相似的结构,进行结构平均,并对模型的几何形状进行规范化处理。
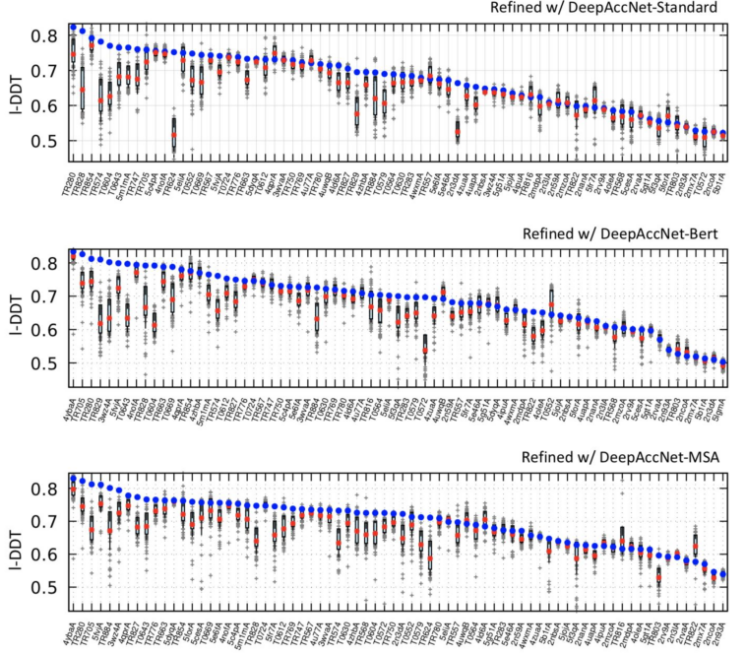
图2.使用DeepAccNet-Standard、-Bert和-MSA最终选择的模型质量
3.结果
DeepAccNet网络结构在给定蛋白质结构模型的基础上做出了三种类型的预测:反映局部结构准确性的每个残基Cβ的局部距离差异测试(Cβl-DDT)分数、阈值为15 Å的Cβ接触图(masks),以及与相应的原生结构相比有符号的Cβ-Cβ距离误差的残基对分布(estograms)。文章指出,DeepAccNet不是预测每对位置的单个误差值,而是预测误差直方图,该直方图提供了有关可能结构分布更为详细的信息,并能够更好的表示误差预测所固有的不确定性。下图展示了没有MSA或Bert嵌入的DeepAccNet对三个靶蛋白(3lhnA、4gmqA和3hixA)中每个蛋白的两个随机选择的诱饵结构的预测示例。在每一种情况下,网络为两个诱饵生成不同的有符号的残基-残基距离误差图,同时也准确预测了不同诱饵的残基模型精度(Cβl-DDT分数)的变化。从图中可以看出,4gmqA的左样本比其他样本更加接近原生结构,网络正确预测了较小误差的位置,并且Cβ l-DDT分数更接近1。总的来说,虽然预测并不完美,但是预测结果提供了很多信息,包括结构的哪些部分需要移动以及以何种方式引导优化。
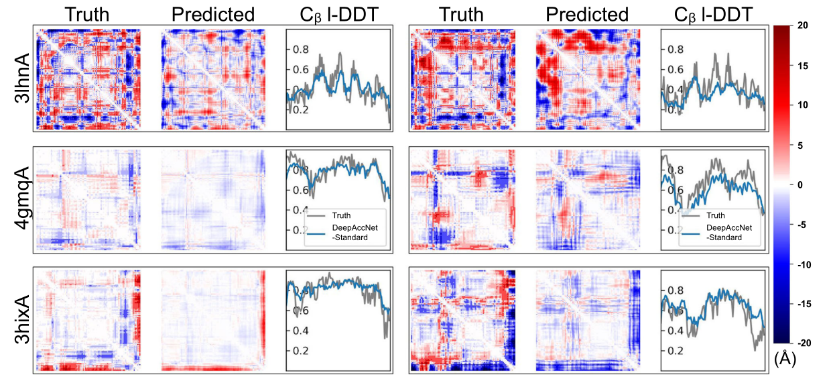
图3.DeepAccNet-Standard在三个靶蛋白随机诱饵结构上的预测示例
文章将DeepAccNet网络的性能与仅根据残基-残基Cβ距离训练的基线网络的性能进行了比较,对于几乎所有的测试集蛋白质,DeepAccNet的平均表现都要好很多。对于所有网络,在Cβ l-DDT分数的预测性能方面并没有随着规模的增加而大幅下降,但在estogram预测性能方面,当蛋白质规模较大时,网络性能有着明显下降。这是因为对于具有更多长距离相互作用的大型蛋白质来说,估计错误的方向和大小是一项更难的任务,而由于Cβ l-DDT分数只考虑短距离的局部变化,它们随着规模的增加降低的较少。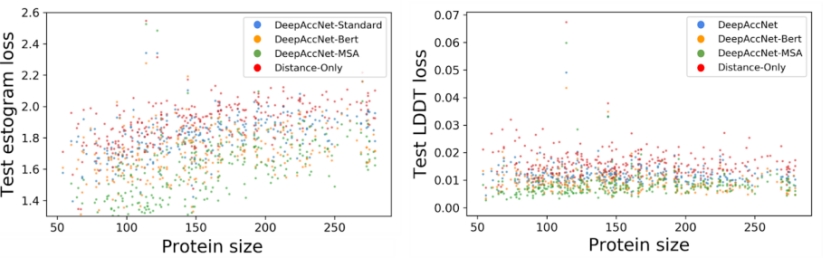
图4.蛋白质规模对网络预测性能的影响
除了距离图特征外,DeepAccNet网络还有以下输入参数:氨基酸的特性和性质、每个残基的局部原子3D环境、主链扭角和残基-残基方向、Rosetta能量项、二级结构信息、MSA以及Bert信息。为了研究这些特征对网络性能的贡献,在训练期间将每一个特征与距离图结合,并通过estogram交叉熵损失和Cβ l-DDT分数均方差在测试集上评估性能。除了MSA特征,最大的贡献来自于基于三维卷积特征和Bert嵌入。
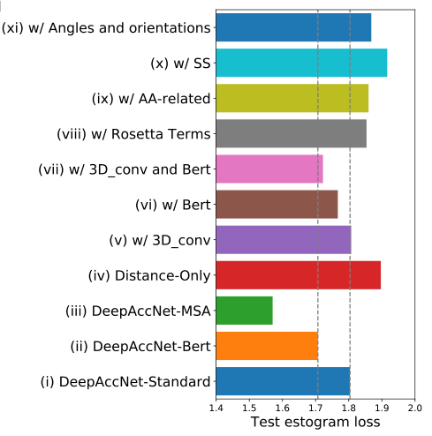
图5.各特征对于网络性能的贡献
一种有效的精度预测方法应该有助于评估和识别实验确定的结构和计算模型中的潜在误差。文章研究了网络对X射线晶体学、核磁共振波谱学(NMR)和电子显微镜(EM)所确定的实验结构的性能表现,结果显示对于高分辨晶体结构,DeepAccNet预测的Cβ l-DDT分数接近于1.0,而对于较低分辨率的结构预测结果有所下降。
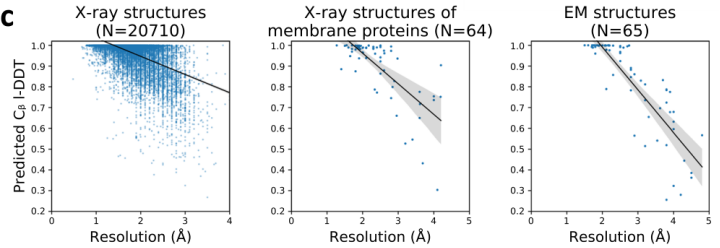
图6.DeepAccNet对于不同分辨率晶体结构的性能表现
从最近的CASP实验中可以清楚看出,从多序列比对中获得的协同进化信息提供了详细的结构信息,将其作为DeepAccNet可选的输入,这样做的原因有两个:首先,所有可用的同源和共同进化信息通常已经用于生成蛋白质结构优化的输入模型;其次,在一些应用中不存在进化多序列比对信息。DeepAccNet-Bert包含Bert嵌入,它使用单个序列生成,没有任何进化比对。在没有同源序列信息的蛋白质EMA任务上(下图虚线左侧),DeepAccNet-Bert的表现优于DeepAccNet-MSA;当有多序列比对信息时(下图虚线右侧),DeepAccNet-MSA是一个更为可靠的选择。
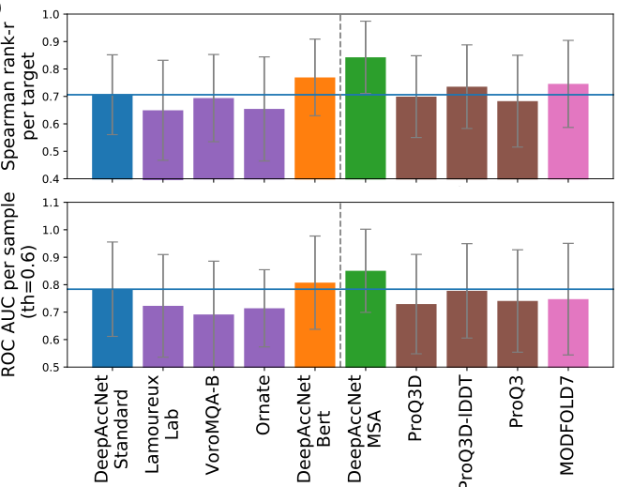
图7.不同EMA方法在CASP13数据上的性能比较文章将网络准确性预测纳入了Rosetta细化协议。Rosetta的高分辨率精化从单个模型开始,在第一个多样化阶段使用一组采样算子探索周围的能量图景,然后在随后的迭代强化阶段专注于空间的最低能量区域。搜索利用一种进化算法,通过多次迭代来保持多样但低能量池。原始协议在输入模型周围采样以保持搜索集中在空间的相关区域,这种做法因限制了搜索区域而有着明显缺点。
为指导搜索,对estograms和Cβ l-DDT分数进行预测,并在Rosetta细化协议的每一次迭代中纳入了三个层次。首先,estograms转换为残基-残基相互作用势,每一对的权重由estograms预测置信度的函数决定,这些势能添加到Rosetta能量函数中作为约束来指导采样;其次,每个残基的Cβ l-DDT预测用于决定哪些区域集中采样或与其他模型重组;第三,全局Cβ l-DDT预测作为进化算法选择阶段的目标函数,并在迭代过程中控制模型在池中的多样性。
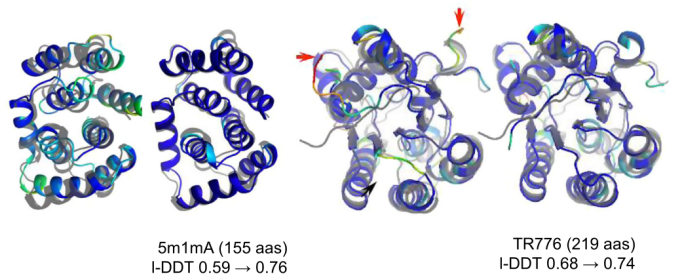
通过基准测试发现,基于网络的精度预测不断提高了基准示例的细化。由于蛋白质的大小,许多基准集中蛋白质的细化在以前是相当具有挑战性的,然而利用更新后的协议,无论蛋白质的大小如何,对起始模型和无指导搜索模型都能观察到一致改进。通过回溯细化轨迹发现,预测和实际模型质量的进展都是在各个阶段逐步发生的,并且两者是相关的。由下图可以看出每个残基模型质量的预测也与实际值吻合得很好。
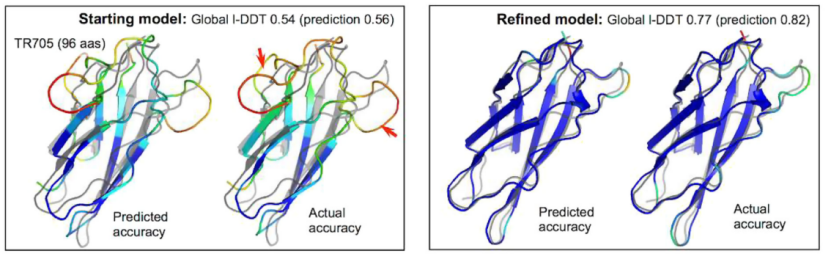
图8.模型在残基预测精度上的改进
预测模型的改进包括在整体结构正确时识别和修改错误区域,以及在模型的核心部分有些不准确时进行整体协调运动。精度预测网络从两个方面促进了这一整体改进:第一,它在每个可以集中采样的模型的每次迭代中提供了更准确的不可靠距离对和区域的估计,第二,它提供了一种方法,通过残基-残基对约束,在已经精确建模的区域中有效地约束搜索空间。
总结
本文工作中,作者提出了DeepAccNet框架,DeepAccNet对以残基为中心的原子坐标进行3D卷积,将详细的残基信息与其他单独的残基以及残基对的信息集成在一起。实验结果表明, DeepAccNet可以提供先进的蛋白质模型精度预测,并且可以将它们进一步用于预测带符号的距离误差,指导蛋白质结构模型优化。









