摘要:预测分子结构和气味之间的关系仍然是一项长达数十年的艰巨任务。这个问题被称为定量结构-气味关系(QSOR)建模,在化学、影响人体营养、合成香料制造、环境和感官神经科学方面是一个重要的挑战。我们提出使用图神经网络进行QSOR,并在一个由嗅觉专家标记的新数据集上显示了它们的显著性能。另外的分析表明,从图神经网络中学习的嵌入捕捉到了结构和气味之间潜在关系的有意义的气味空间表示,在两个具有挑战性的迁移学习任务上的出色表现证明了这一点。机器学习已经对视觉和听觉产生了巨大的影响。基于这些关于分子特性的图形神经网络的早期结果,我们希望机器学习最终能够在嗅觉方面起到它已经在视觉和听觉上所做的作用。
预测分子的性质是机器学习中一个日益增长的研究领域[1,2],特别是随着从图值输入中学习的模型的复杂性和稳健性提高[3,4]。在研究活动激增期间,一个相对较少受到关注的分子性质预测问题是建立定量结构-气味关系(QSOR)模型(与药物化学中的定量结构-活性关系相反)。这是一个跨越化学、物理、神经科学和机器学习的70多年前的问题[5]。
人类对气味的感知是300-400种不同类型嗅觉受体(ORs)激活的结果,这些ORs表达在数百万个嗅觉感觉神经元(OSN)中,嵌入在一小块5 cm2的称为嗅觉上皮的组织中。这些OSN将信号发送到嗅球,然后发送到大脑中的其他结构[6,7]。视觉和听觉深度学习的进展表明,我们或许能够直接预测输入刺激的最终感觉结果。嗅觉深度学习的进展将有助于发现新的合成气味,从而减少寻找天然产品对人嗅觉的影响。此外,从气味识别任务训练的模型中获得的分子的新表达方式可能有助于我们理解大脑中的感官知觉[8]。
在这里,我们整理了一个与专家标记的气味描述符相关的分子数据集(在QSOR中,气味描述符指的是我们希望预测的性质,而不是它们在化学信息学中的使用,在化学信息学中,它们指的是模型的输入特征)。我们训练图形神经网络(GNNs)[4,9],仅用分子的图形结构来预测这些气味描述符。我们表明,我们的模型学习了气味空间的表示,它基于感知相似性而不是纯粹的结构相似性,在全球和局部尺度上对分子进行聚类。此外,我们还表明,这种表示对于预测相关任务是有用的,这是机器学习在化学应用中的一个发展领域[10,11]。这些结果表明,我们的建模方法捕捉到了分子结构和气味之间关系的通用表示,我们预计这对于合理的分子设计和筛选是有用的。
QSOR的问题是古老的[12],但在科学文献中第一次出现是Amoore、Schiffman和Dyson等人[13, 14, 15]发表的一篇文章。现代人试图以直接的数据驱动和统计学的方式来解决这个问题的尝试始于几十年前[5],甚至包括神经网络的早期应用[16]。然而,在这些早期的研究中,所使用的气味描述符的数量很少(少于十个,通常是一个),而且总的刺激物的数量也很有限(通常是10个,很少是100个分子)[17, 18, 19]。由于其难度,这个问题长期以来一直是一个悬而未决的问题--分子结构的极小变化就会对其气味产生巨大的影响,这种现象在药物化学中被称为活性悬崖[20, 21]。一个典型的例子是新铃兰醛(Lyral),它是一个商业上成功的分子,闻起来有铃兰muguet的味道(一种经常用于烘干机片的花香)。它的结构上的近邻并不总是感知上的近邻,而且它的一些感知上的近邻在结构上的相似性很小(图1)。
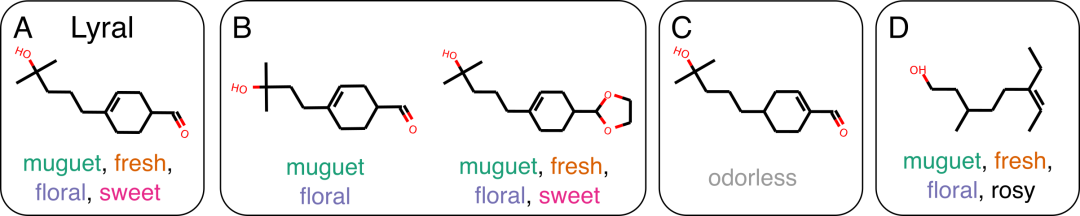
图1:结构相似的分子不一定有相似的气味描述符。A.新铃兰醛,参考分子。B. 具有相似结构的分子可以共享相似的气味描述符。C. 然而,一个很小的结构变化就可以使分子变得无味。D.此外,较大的结构变化可以使分子的气味基本保持不变。引自Ohloff,Pickagen和Kraft[28]。
最近,DREAM嗅觉挑战赛刺激了传统机器学习方法在QSOR预测中的应用[22]。这项挑战提出了一个数据集,49位未经训练的小组成员对476个分子的21个气味属性进行了模拟评分。DREAM挑战赛的获胜模型主要依靠Dragon分子特征[23]或Morgan指纹[24]作为分子的特征化。这些特征被随机森林用来进行预测,这是一种在化学信息学中有着长期成功记录的方法。我们在这项工作中使用这些方法作为基线。
我们希望强调一些现代机器学习的QSOR方法。Tran及其同事[25]重新审视了神经网络在这项任务中的应用,并开发了一个卷积神经网络,将分子的自定义三维空间表示作为输入。Nozaki等人[26]利用分子的质谱和自然语言处理工具来预测气味的文本描述。Gutierrez等人[27]使用分子的词嵌入和化学信息学表征来预测气味特性。
2.1 对分子进行特征化处理并对其属性进行建模的经典方法
QSOR在历史上使用了许多来自化学信息学和药物化学的计算技术。为了预测分子特性,通常使用手工制作的特征将分子转化为固定长度的向量,并将其输入预测模型,如随机森林或完全连接的神经网络[2, 29]。我们在下面描述了分子特征化的基准方法的细节。
Dragon and Mordred Features
有几种可用的手工制作的分子特征,这在嗅觉神经科学领域很受欢迎。Dragon(闭源,Mauri2006-sp)和Mordred(开源,Moriwaki2018-hv)都是包含数千个计算分子特征的方法。它们是几种类型的分子信息和统计数据的集合,例如原子类型计数、图拓扑统计和酸/碱计数。其中一些特征很容易解释(例如碳原子的数量),而另一些则不然(例如距离/绕行矩阵的4阶谱矩)。我们在目前的工作中使用了Mordred,因为它是开源的,并且我们发现它和Dragon之间在预测性能上没有明显的差异(数据未显示)。
Molecular Fingerprints
分子指纹将分子图的拓扑环境编码为固定长度的向量。环境是分子图的一个片段,表示存在单一原子类型或官能团,例如醇或酯基。这种表征分子的方法在药物化学领域很受欢迎;传统上,基于比特的摩根指纹被用于化学信息学,利用谷本相似度Maggiora2014-va来检索最近邻的分子。当这些环境以原子为中心并通过相邻原子构建时,它们被称为扩展连接指纹,或摩根指纹(rogers - 2010-wo),当它们通过图中的路径构建时,它们被称为路径描述符指纹(Randic1999-fn)。更常用的比特变体记录了给定环境的存在(例如,这个分子中是否存在酯?),而计数变体记录了给定环境的实例数量(例如,这个分子中有多少个酯基?)。这个信息被散列成一个固定长度的向量。有两个可调参数:最大拓扑半径和指纹矢量大小。最大拓扑半径决定了指纹可以表示的最大碎片。指纹向量大小控制尖锐冲突的可能性。我们调整这两个参数以最大化预测性能。
在我们的基线实验中,我们明确比较了基于比特的路径描述符指纹(bFP)和基于计数的摩根指纹(cFP)。使用化学信息学软件包RDKit生成两种类型的指纹。分子特性通常使用随机森林或支持向量机等模型进行预测,因此我们使用随机森林作为每个bFP和cFP特征的预测模型。
大多数机器学习模型需要规则形状的输入(例如像素网格或数字向量)作为输入。最近,图神经网络(GNN)已经能够将不规则形状的输入(如图形)直接用于机器学习应用[Wu2019]。使用领域包括预测社交网络图中的友谊,学术文献中的引用网络,以及与本工作最相关的化学分类和回归任务[2018-yg]。3.1GRAPH NEURAL NETWORKS FOR PREDICTING MOLECULAR PROPERTIES
通过把原子看作节点,把化学键看作边,我们可以把分子看作一个图。gnn是节点和边的可学习置换不变变换,它产生固定长度的向量,由全连接神经网络进一步处理。与专家精心制作的通用特征相比,gnn可以被认为是专门用于任务的可学习特征器。Duvenaud2015-ye。gnn在预测分子Wu2018-yg的生物物理、生物、物理和电子量子特性方面取得了最先进的成果,因此我们相信它们在QSOR中的应用是有希望的。
GNN由消息传递层组成,每个层都有一个约和操作,然后是几个完全连接的层。架构细节可以在附录中找到,表超参数调优和GNN架构。最终的完全连接层的输出数量等于被预测的气味描述符的数量。图2说明了我们的模型。我们使用TensorFlow软件包abadi2016tensorflow实现这些GNN模型。
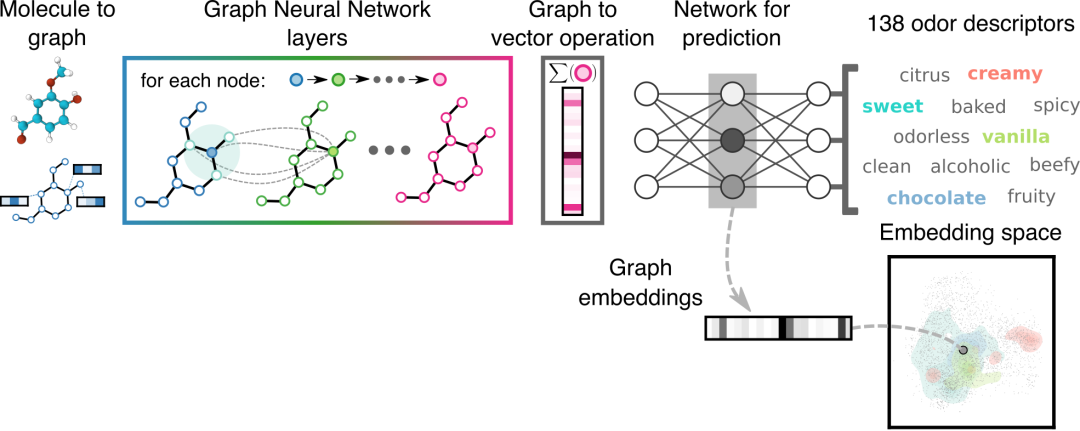
图2:模型示意图。每个分子首先以其组成原子、键和连通性为特征。每个图神经网络(GNN)层,在这里用不同的颜色表示,转换前一层的特征。最终GNN层的输出被简化为一个向量,然后通过一个全连接的神经网络用于预测气味描述符。我们从模型的倒数第二层检索图嵌入。右下角显示了四个气味描述符的嵌入空间表示的示例;该图中区域的颜色与右上方气味描述符的颜色相对应。
3.2LEARNED GRAPH NEURAL NETWORK EMBEDDINGS
所有深度神经网络架构都在中间层构建输入数据的表示。深度神经网络在预测任务中的成功依赖于其学习表征的质量,通常被称为嵌入。例如,ImageNet嵌入通常按原样使用,以对不相关的图像任务进行预测。随着BERT模型及其近亲的出现,这种使用预训练嵌入的能力在自然语言处理Devlin2018-rp中变得越来越普遍。学习嵌入的结构甚至可以导致对任务或问题领域的见解,嵌入甚至可以成为研究对象。coenen2019visualizing。
我们将倒数第二个完全连接层的激活保存为固定维度的“气味嵌入”。GNN模型必须将分子的图结构转换为对分类有用的固定长度表示。尽管学习神经网络嵌入分子的应用还很年轻,而且相对未经证实。Zhavoronkov2019-pw,我们仍然期望在气味预测任务上嵌入学习到的GNN可能包括语义上有意义和有用的气味分子组织。我们在本工作的后面部分明确测试了这种气味嵌入的效用。
4 一个富有成效的QSOR数据集
我们从两个不同的来源收集了5030个经过专家标记的分子:GoodScents香水材料数据库(n=3786, noauthor_unddate -uu)和Leffingwell PMP 2001数据库(n=3561, Leffingwell2005-uv)。数据集共享2317重叠的分子。嗅觉专家(通常是执业调香师)用一个或多个气味描述符标记分子,从而产生多标签预测问题。GoodScents为每个分子描述了1-15个气味描述符(图3A),而Leffingwell使用自由格式文本。使用goodscent本体对气味描述符进行规范化,重叠分子继承了两个数据集气味描述符的联合。在过滤了至少有30个代表性分子的气味描述符后,剩下138个气味描述符(图3B),其中包括一个无气味描述符。有些气味描述符非常常见,比如水果味或绿色味,而其他气味描述符则很少见,比如萝卜味或面包味。该数据集由香水材料组成,因此偏向于异味化合物。由于不同的特异性水平,标签计数也会出现偏差,例如水果味总是比菠萝味更常见。
气味描述符之间存在非常强的共现结构,反映了气味描述符相似和不相似的常识性直觉(图3C)。例如,有一个乳制品集群,其中包括乳制品、酸奶、牛奶和奶酪描述符,表明它们经常作为描述符共同出现在单个分子中。还有一个水果集群,有苹果、梨、菠萝等,还有一个面包房集群,包括烤、坚果和可可等。以前的QSOR方法通常为每个气味描述符训练一个模型。为了利用这种相关结构,我们应用GNNs一次预测所有138个气味描述符任务。
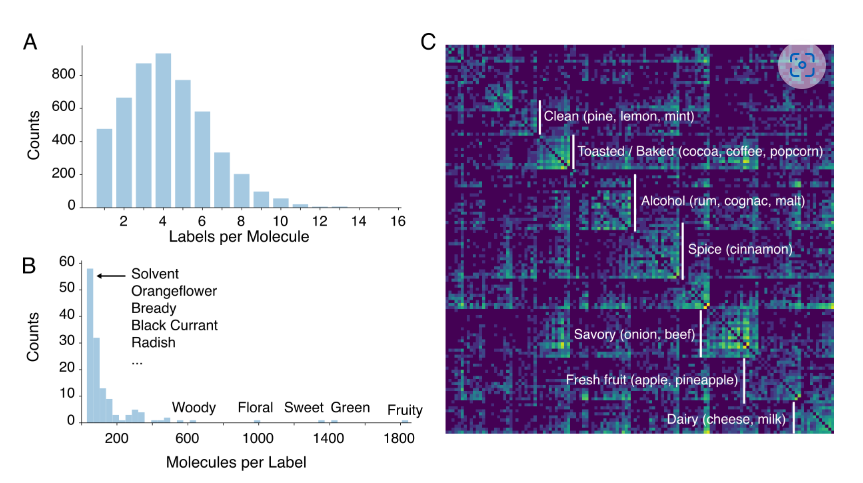
图3:数据集概述。A.气味描述符频率分布。B.标签密度分布。气味描述符的共现矩阵。10个最常见的描述符被移除以保持视觉清晰度,剩余的描述符使用光谱聚类重新排序。突出显示了主要气味组及其示例。颜色范围是在对数尺度上的,并且标准化使得每行和每列之和为1。
5 QSOR预测性能基准
我们将数据集中每个气味描述符的分类性能作为一个多标签分类问题进行基准测试。我们将GNN模型与随机森林模型(RF)和k近邻模型(KNN)在基于位的RDKit指纹(bFP)、基于计数的Morgan指纹(cFP)和Mordred特征上进行了比较。我们报告了几个指标(表1),因为每个指标可以突出不同的性能特征。对于其余的分析,我们主要比较模型的平均AUROC,平均气味描述符;按标签划分的AUROC性能如图4所示。我们在cFP和bFP特征上训练了非基于图的全连接神经网络,但它们的性能与RF模型没有区别(数据未显示)。
表1:气味描述符预测结果。平均值,95% CI[下界,上界]。报告的数字是所有138个气味描述符的未加权平均值;见补充表每描述符结果表的气味标签报告的结果。准确率/召回率决策阈值针对从训练集创建的交叉验证分割上的F1分数进行了优化。每个指标的最佳值以粗体显示。召回率没有统计上的赢家,因此省略;这些得分范围为0.365-0.393,所有模型之间有很高的重叠。
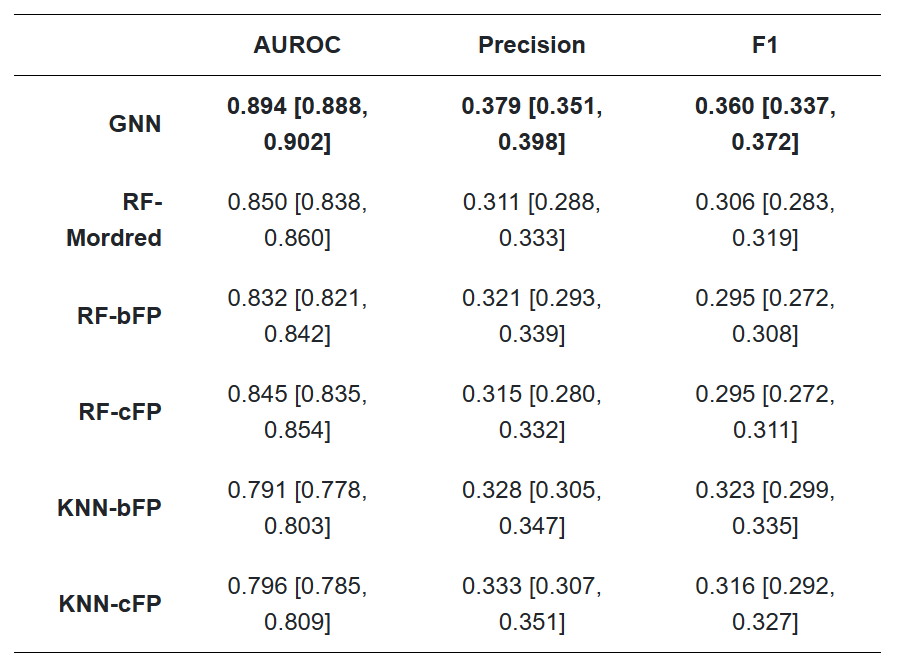
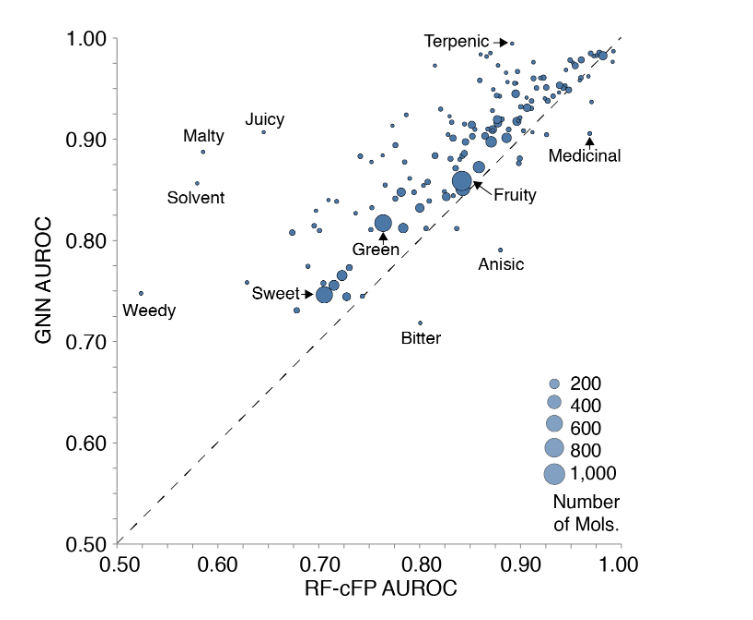
图4:RF-cFP和GNN的比较,按气味描述符分解。每个点代表一个气味描述符,大小代表正面例子的数量。GNN在几乎所有气味描述符上都优于RF-cFP。
6 评估气味嵌入
反映气味之间常识性关系的气味嵌入应该具有全局和局部的结构。具体来说,对于全局结构,感知上相似的气味应该在嵌入的附近。对于局部结构,具有相似气味感知的单个分子应该聚在一起,从而在嵌入中靠近。我们依次考察这两个性质。
6.1 检查一个习得的气味空间的整体结构
我们从训练好的GNN模型的倒数第二层输出中获取每个数据点的嵌入表示。在我们最好的模型中,每个分子都被映射到一个63维的向量上。定性地,为了在二维中可视化这个空间,我们使用主成分分析(PCA)来降低它的维数。使用核密度估计(KDE)可以突出显示共享相似标签的所有分子的分布。
嵌入空间的全局结构如图5所示。在这个例子中,我们发现单个气味描述符(如麝香、卷心菜、百合和葡萄)倾向于在它们自己的特定区域聚集。对于经常出现的气味描述符,我们发现嵌入空间捕获了气味描述符中隐含的层次结构。气味标签茉莉,薰衣草和木桂花的集群被发现在更广泛的气味标签花的集群。如果我们检查我们学习到的嵌入中所有气味之间的两两距离,我们可以看到图3C中明显的块结构被学习到的GNN嵌入反映出来,但没有分子指纹(图S1)。此外,降维的分子指纹嵌入不具有相同程度的组织和可解释性(图S3)。
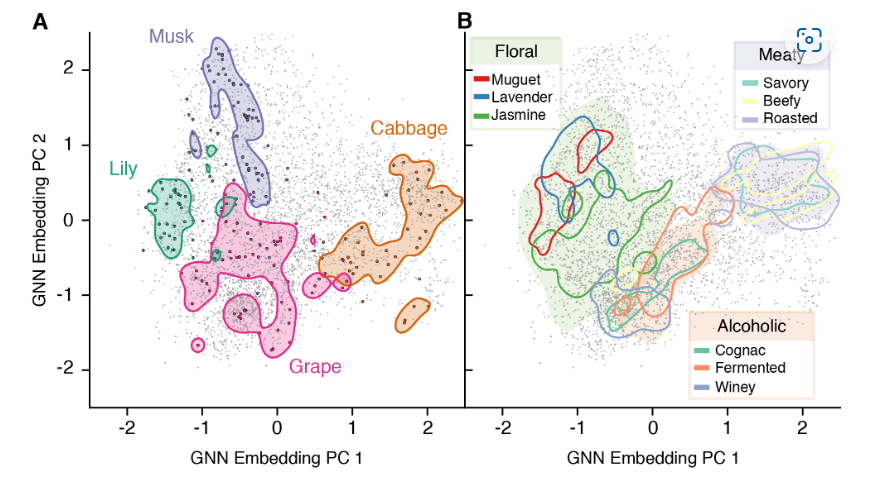
图5:作为学习气味空间的GNN模型嵌入的二维表示。分子被表示为单个点。阴影和轮廓区域是标记数据分布的核密度估计。A.四个低共现的气味描述符在嵌入空间中的重叠度很低。三种一般的气味描述(花香、肉味、酒精味)在它们的范围内都包含了更具体的标签。分子指纹等效分析见补充图S3。
6.2 评估学习到的气味空间的局部结构
我们测试了嵌入空间附近的分子是否具有感知相似性。具体来说,我们询问在我们的GNN嵌入中具有小余弦距离的分子是否在感知上相似。我们使用谷本距离作为基线,它相当于bFP特征上的Jaccard距离。谷本距离是化学信息学中常用的分子数据库查找度量。然而,具有相似结构特征的分子并不总是气味相同(图1),因此我们预计使用bFP特征的近邻可能在感知上不像使用我们的嵌入的邻居那样相似。
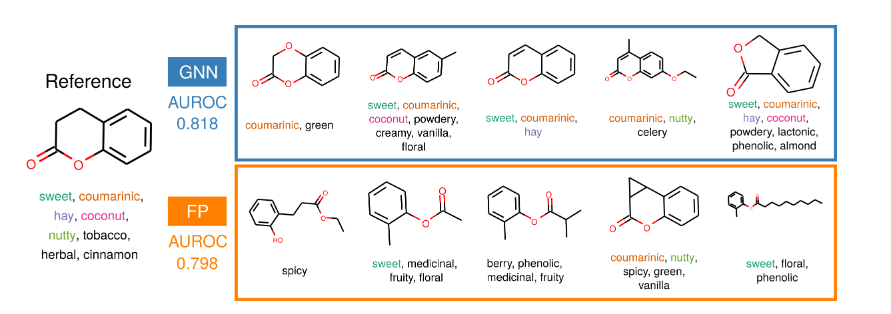
图6:最近邻检索。显示了GNN嵌入上的余弦相似度和基于位的摩根指纹上的谷本距离的前五个最近邻。图中显示了k-最近邻分类器(k=20)在所有气味描述符上的平均性能,并使用相应的特征表示进行了训练。
我们训练了一个k近邻(KNN)分类器(k=20)从GNN嵌入和bfp中预测气味描述符。GNN嵌入(AUROC = 0.818, 95% CI[0.806, 0.830])优于bFP (AUROC = 0.782, 95% CI[0.773, 0.797])。检查每种方法找到的最近邻居(图6)表明,两种方法都产生具有相似结构特征的分子,但使用GNN嵌入的检索产生的分子在感知上与源分子更相似。这表明我们的表征比基于比特的指纹更能通过气味感知相似性来聚类分子。图S2和表S4显示了使用bFPs和GNN嵌入的不同距离度量比较分子之间气味感知相似性和嵌入距离的其他结果。
我们已经证明,我们的嵌入空间具有全局和局部结构,反映了气味描述符的常识和心理物理组织。在下面的部分中,我们将展示这种组织是有用的,并且这种嵌入可以用于对相邻的、具有挑战性的任务进行预测。
6.3 将学习转移到以前看不见的气味描述符
气味描述符可能是新发明或改进的(例如,具有梨描述符的分子可能后来归属于更具体的梨皮、梨茎、梨肉、梨核描述符)。一个有用的气味嵌入可以将学习到的Pan2010-jk转移到这个新的描述符上,只使用有限的数据。为了近似这个场景,我们每次从数据集中删除一个气味描述符。使用从(N−1)气味描述符中训练的嵌入作为特征,我们训练了一个随机森林来预测先前的气味描述符。我们使用cFP和Mordred特征作为比较的基线。结果如图7所示。在这个任务中,GNN嵌入显著优于Morgan指纹和Mordred特征(无重叠95%置信区间,Mordred结果见表1),但正如预期的那样,仍然比在目标气味上训练的GNN表现略差。这表明基于gnn的嵌入可以推广到预测新的但相关的气味。
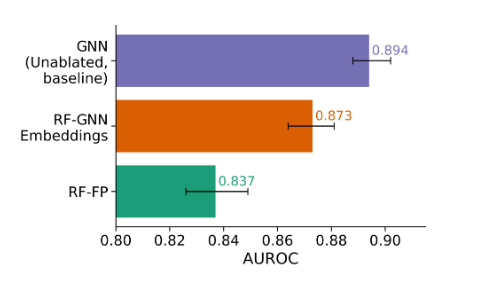
图7:先前残留气味的平均AUROC。单标签消融任务中所有标签的平均AUROC评分。误差条表示95%置信区间。顶部的条形图表示模型在所有标签上训练的性能。中间的条形图表示随机森林模型的性能,该模型使用在(N−1)个气味标签上训练的模型的GNN嵌入进行训练。底部的条形图表示经过摩根指纹计数训练的随机森林。
6.4 推广到其他嗅觉任务:未知嗅觉预测挑战
DREAM嗅觉预测挑战赛Keller2017-vs是一项公开竞赛,在未经训练的小组成员收集的数据集上构建QSOR模型。DREAM数据集与我们自己的数据集有几个不同之处。首先,这是一个回归问题——小组成员在1到100的范围内对一个分子闻到特定气味描述符的数量进行了评级。第二,它有476个分子~ 5k(尽管我们的数据集包含了几乎所有的DREAM分子)。第三,评级是由一大批未经训练的个人在短时间内提供的,而我们的评级是由一小部分专家在多年内收集的。DREAM挑战通过模型预测与每个气味描述符的平均报告强度的Pearson’s r相关性来测量模型性能,如图8所示。图S4、S5中显示了R2和95%置信区间等其他统计数据。
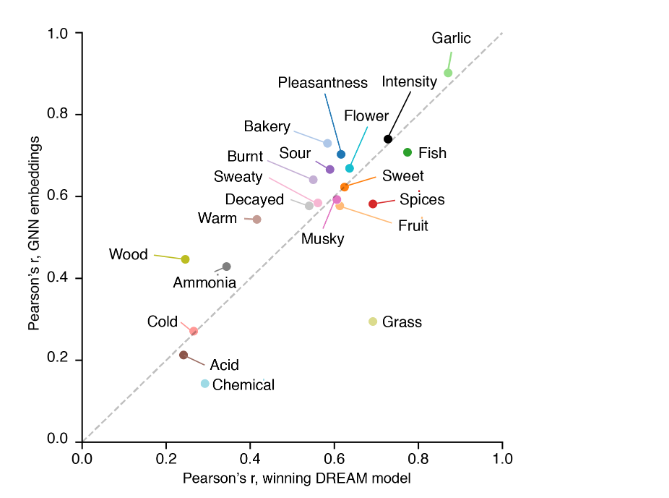
图8:GNN和最佳基线模型在DREAM嗅觉预测挑战中的预测性能。皮尔逊的r代表DREAM挑战的获胜者(对于大多数气味,龙特征子集上的随机森林)与训练在GNN嵌入上的RF(按气味描述符分解)。灰色虚线表示两种模型的性能相同。在对角线以上的数据点表示更好的GNN预测性能。虽然GNN模型的平均值通常较高,但最先进的模型和GNN在统计上都无法区分。
获胜的DREAM模型使用了随机森林模型,结合了几种特征来源,主要是Dragon和Morgan指纹,以及其他信息来源。仅使用我们与调谐随机森林模型的嵌入,我们就获得了平均皮尔逊的r=0.55,而上述最先进的模型实现了平均皮尔逊r=0.54。虽然我们能够在13个任务中有更好的平均表现,但当考虑置信区间时,我们发现两个任务的表现是无法区分的r和R2回归得分(图S4, S5)。
总的来说,这表明我们的QSOR建模方法可以推广到相邻的感知任务,并捕获有关人类嗅觉感知的有意义和有用的结构,即使在不同的背景下,使用不同的方法进行测量。
7 结论
我们收集了大量经过专业标记的单分子气味剂的新数据集,并训练了一个图神经网络来预测分子结构与其气味之间的关系。我们展示了关于现场识别基线的最先进的QSOR任务结果。此外,我们发现嵌入在局部和全局尺度上都捕获了有意义的结构。最后,我们证明了我们的模型学习的嵌入在下游任务中是有用的,这是目前现代机器学习模型和化学数据中罕见的特性。因此,我们相信我们的模型及其学习嵌入可能在合理设计新气味剂方面普遍有用。
8 致谢
我们感谢D. Sculley, Steven Kearnes, Jasper Snoek, Emily Reif, Carey Radebaugh和David Belanger对手稿的支持,建议和有用的讨论。我们感谢Aniket Zinzuwadia的讨论,他为他的毕业论文做了DREAM和GoodScents数据集的初步工作。我们感谢GoodScents的Bill Luebke和Leffingwell and Associates的John Leffingwell慷慨地分享他们的数据用于研究。我们感谢谷歌大脑剑桥办公室的所有同事创造和维护这样一个支持和激励的环境。
参考文献
[1] Zhenqin Wu, Bharath Ramsundar, Evan N Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S Pappu, Karl Leswing, and Vijay Pande. MoleculeNet: a benchmark for molecular machine learning, 2018.
[2] John B O Mitchell. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci., 4(5):468–481, September 2014.
[3] Franco Scarselli, Marco Gori, Ah Chung Tsoi, Markus Hagenbuchner, and Gabriele Monfardini. Computational capabilities of graph neural networks. IEEE Trans. Neural Netw., 20(1):81–102, January 2009.
[4] Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl. Neural message passing for quantum chemistry. April 2017.
[5] Karen J Rossiter. Structure-odor relationships. Chem. Rev., 96(8):3201–3240, 1996.
[6] Chih-Ying Su, Karen Menuz, and John R Carlson. Olfactory perception: receptors, cells, and circuits. Cell, 139(1):45–59, October 2009.
[7] John P McGann. Poor human olfaction is a 19th-century myth, 2017.
[8] Daniel L K Yamins and James J DiCarlo. Using goal-driven deep learning models to understand sensory cortex, 2016.
[9] David K Duvenaud, Dougal Maclaurin, Jorge Iparraguirre, Rafael Gómez-Bombarell, Timothy Hirzel, Alán Aspuru-Guzik, and Ryan P Adams. Convolutional Networks on Graphs for Learning Molecular Fingerprints. In Advances in Neural Information Processing Systems, pages 2215–2223, 2015.
[10] Han Altae-Tran, Bharath Ramsundar, Aneesh S Pappu, and Vijay Pande. Low data drug discovery with One-Shot learning. ACS Cent Sci, 3(4):283–293, April 2017.
[11] Clyde Fare, Lukas Turcani, and Edward O Pyzer-Knapp. Powerful, transferable representations for molecules through intelligent task selection in deep multitask networks. September 2018.
[12] Charles Sell.
Perfume in the Bible. Royal Society of Chemistry, July 2019.
[13] J E Amoore, J W Johnston, Jr, and M Rubin. THE STEROCHEMICAL THEORY OF ODOR. Sci. Am., 210:42–49, February 1964.
[14] G Malcolm Dyson. Raman effect and the concept of odour. Perfum. Essent. Oil Rec, 28:13, 1937.
[15] Susan S Schiffman. Physicochemical correlates of olfactory quality. Science, pages 112–117, 1974.
[16] M Chastrette, D Cretin, and C el Aïdi. Structure-odor relationships: using neural networks in the estimation of camphoraceous or fruity odors and olfactory thresholds of aliphatic alcohols. J. Chem. Inf. Comput. Sci., 36(1):108–113, January 1996.
[17] Sigma-Aldrich Corporation. Aldrich Chemistry 2012-2014: Handbook of Fine Chemicals. 2011.
[18] Andrew Dravnieks and ASTM Committee E-18 on Sensory Evaluation of Materials and Products. Section E-18.04.12 on Odor Profiling. Atlas of odor character profiles. Astm Intl, 1985.
[19] Steffen Arctander. Perfume and flavor chemicals:(aroma chemicals), volume 2. Allured Publishing Corporation, 1969.
[20] C S Sell. On the unpredictability of odor, 2006.
[21] Dagmar Stumpfe and Jürgen Bajorath. Exploring activity cliffs in medicinal chemistry. J. Med. Chem., 55(7):2932–2942, April 2012.
[22] Andreas Keller, Richard C Gerkin, Yuanfang Guan, Amit Dhurandhar, Gabor Turu, Bence Szalai, Joel D Mainland, Yusuke Ihara, Chung Wen Yu, Russ Wolfinger, Celine Vens, Leander Schietgat, Kurt De Grave, Raquel Norel, DREAM Olfaction Prediction Consortium, Gustavo Stolovitzky, Guillermo A Cecchi, Leslie B Vosshall, and Pablo Meyer. Predicting human olfactory perception from chemical features of odor molecules. Science, 355(6327):820–826, February 2017.
[23] Andrea Mauri, Viviana Consonni, Manuela Pavan, and Roberto Todeschini. Dragon software: An easy approach to molecular descriptor calculations. Match, 56(2):237–248, 2006.
[24] David Rogers and Mathew Hahn. Extended-connectivity fingerprints. J. Chem. Inf. Model., 50(5):742–754, May 2010.
[25] Ngoc Tran, Daniel Kepple, Sergey A Shuvaev, and Alexei A Koulakov. DeepNose: Using artificial neural networks to represent the space of odorants. November 2018.
[26] Yuji Nozaki and Takamichi Nakamoto. Predictive modeling for odor character of a chemical using machine learning combined with natural language processing. PLoS One, 13(6):e0198475, June 2018.
[27] E Darío Gutiérrez, Amit Dhurandhar, Andreas Keller, Pablo Meyer, and Guillermo A Cecchi. Predicting natural language descriptions of mono-molecular odorants. Nat. Commun., 9(1):4979, November 2018.
[28] Günther Ohloff and Wilhelm Pickenhagen. Scent and Chemistry. Wiley, January 2012.
[29] Vladimir Svetnik, Andy Liaw, Christopher Tong, J Christopher Culberson, Robert P Sheridan, and Bradley P Feuston. Random forest: a classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci., 43(6):1947–1958, November 2003.
[30] Hirotomo Moriwaki, Yu-Shi Tian, Norihito Kawashita, and Tatsuya Takagi. Mordred: a molecular descriptor calculator. J. Cheminform., 10(1):4, February 2018.
[31] Gerald Maggiora, Martin Vogt, Dagmar Stumpfe, and Jürgen Bajorath. Molecular similarity in medicinal chemistry. J. Med. Chem., 57(8):3186–3204, April 2014.
[32] David Rogers and Mathew Hahn. Extended-connectivity fingerprints. J. Chem. Inf. Model., 50(5):742–754, May 2010.
[33] Milan Randić and Subhash C Basak. Optimal molecular descriptors based on weighted path numbers. J. Chem. Inf. Comput. Sci., 39(2):261–266, March 1999.
[34] RDKit: Open-source cheminformatics. http://www.rdkit.org.
[35] Zonghan Wu, Shirui Pan, Fengwen Chen, Guodong Long, Chengqi Zhang, and Philip S. Yu. A comprehensive survey on graph neural networks. CoRR, abs/1901.00596, 2019.
[36] Martín Abadi, Paul Barham, Jianmin Chen, Zhifeng Chen, Andy Davis, Jeffrey Dean, Matthieu Devin, Sanjay Ghemawat, Geoffrey Irving, Michael Isard, et al. Tensorflow: A system for large-scale machine learning. In 12th {USENIX} Symposium on Operating Systems Design and Implementation ({
OSDI} 16), pages 265–283, 2016.
[37] Yoshua Bengio, Aaron Courville, and Pascal Vincent. Representation learning: a review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell., 35(8):1798–1828, August 2013.
[38] Jeff Donahue, Yangqing Jia, Oriol Vinyals, Judy Hoffman, Ning Zhang, Eric Tzeng, and Trevor Darrell. Decaf: A deep convolutional activation feature for generic visual recognition. In International conference on machine learning, pages 647–655, 2014.
[39] Ali Sharif Razavian, Hossein Azizpour, Josephine Sullivan, and Stefan Carlsson. Cnn features off-the-shelf: an astounding baseline for recognition. In Proceedings of the IEEE conference on computer vision and pattern recognition workshops, pages 806–813, 2014.
[40] Jacob Devlin, Ming-Wei Chang, Kenton Lee, and Kristina Toutanova. BERT: Pre-training of deep bidirectional transformers for language understanding. October 2018.
[41] Andy Coenen, Emily Reif, Ann Yuan, Been Kim, Adam Pearce, Fernanda Viégas, and Martin Wattenberg. Visualizing and measuring the geometry of bert. arXiv preprint arXiv:1906.02715, 2019.
[42] Rafael Gómez-Bombarelli, Jennifer N Wei, David Duvenaud, José Miguel Hernández-Lobato, Benjamín Sánchez-Lengeling, Dennis Sheberla, Jorge Aguilera-Iparraguirre, Timothy D Hirzel, Ryan P Adams, and Alán Aspuru-Guzik. Automatic chemical design using a Data-Driven continuous representation of molecules. ACS Cent Sci, 4(2):268–276, February 2018.
[43] Alex Zhavoronkov, Yan A Ivanenkov, Alex Aliper, Mark S Veselov, Vladimir A Aladinskiy, Anastasiya V Aladinskaya, Victor A Terentiev, Daniil A Polykovskiy, Maksim D Kuznetsov, Arip Asadulaev, Yury Volkov, Artem Zholus, Rim R Shayakhmetov, Alexander Zhebrak, Lidiya I Minaeva, Bogdan A Zagribelnyy, Lennart H Lee, Richard Soll, David Madge, Li Xing, Tao Guo, and Alán Aspuru-Guzik. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol., 37(9):1038–1040, September 2019.
[44] The good scents company - flavor, fragrance, food and cosmetics ingredients information. http://www.thegoodscentscompany.com/. Accessed: 2019-9-4.
[45] John C Leffingwell. Leffingwell & associates, 2005.
[46] S J Pan and Q Yang. A survey on transfer learning. IEEE Trans. Knowl. Data Eng., 22(10):1345–1359, October 2010.
[47] Piotr Szymański and Tomasz Kajdanowicz. A network perspective on stratification of Multi-Label data. April 2017.
原文链接:
Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules – arXiv Vanity (arxiv-vanity.com)







